Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Organización, regulación y manipulación genética Dra. Mildred Jiménez
Genética molecular Organización, regulación y manipulación genética Dra. Mildred Jiménez 1

2
Objetivos Comprender las bases moleculares de las mutaciones que conllevan a las enfermedades genéticas Usar tal información para mejorar diagnósticos y tratamientos Avances en nuestro conocimiento de la genética molecular han ayudado considerablemente en el desarrollo de nuevas tecnologías que permiten el análisis detallado de los genes normales y anormales. Hablaremos de técnicas que han sido y continúan siendo las responsables de los avances en la investigación genética tanto básica como aplicada. 2

3
I- Análisis de secuencias individuales de ADN y ARN
3

4
Limitantes iniciales Obtener cantidad suficiente de la secuencia de ADN o ARN de interés a ser analizada. Purificar la secuencia de interés dentro de todos los demás segmentos de ADN o ARNm presentes en la célula. clonaje molecular y PCR Como genetistas moleculares se enfrentan 2 obstaculos fundamentales al llevar a cabo las investigaciones sobre las bases moleculares de las enfermedades hereditarias 1- Ya que cada célula tiene, generalmente, solamente 2 copias de cada gen y además ciertos genes pueden ser que se trasncriban solamente en un grupo de tejidos o solemnete en bajas cantidades o ambos casos, produciendo de esta forma solamente un pequeño número de moléculas de ARNm. Para resolver ambos problemas, durante las últimas 3 décadas han sido testigas de una revolución tecnológica en vistas a resolver tanto los problemas de cantidad y de purificación. Hay que protegar el AND si lo queremos estudiar, concentrarlo para luego estudiarlo. 4

5
Clonaje Molecular Involucra la transferencia de una secuencia de ADN de interés a una célula o microorganismo. Proceso que involucra… Clonaje: copia fiel idéntica, de lo que originalmente se tenía. En una región determinada de un gen uno copia esa región, pero para esto se ocupa un vector, un vector para introducir la secuencia que quiero clonar para luego amplificarla. 5

6
Cultivo del microorganismo
Reproducción de la secuencia de ADN introducida + ADN propio Aislamiento de las secuencias de interés en grandes cantidades Es más fácil duplicar una bacteria que si tenemos una células eucariota. 6

7
Se usan bacterias, estas tienen un plasmido que tienen una repliación apart de la bacteria. Estos plásmidos se puede introducir partes o quitar otras sin afectar a la bateria.

8
Los plásmidos tienen un inicio de replicación.

9
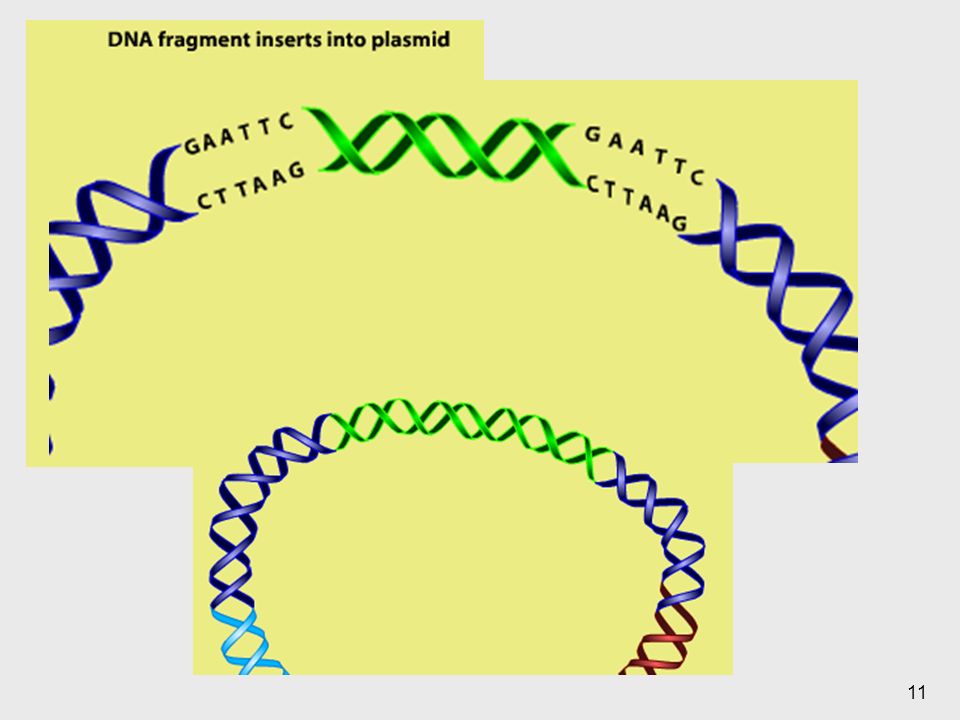
Existe lo que se llaman enzimas de restricción que son aquellas que reconocen un área específica. A la hora de reconocer la región generan un corte para un sitio específico, por lo que se me rompe la doble hélice. Se liberan regiones y va a haber un extremo pegajoso es decir un extremo 3’.

10
Luego de que tengo la región y el plásmido cortado, el ADN va a buscar la región complementaria y luego se da la unión.

12
Esto es un plásmido recombinante, este plasmido será nuestro vector

13
Se polariza un poco la membrana de manera que se abran algunos canales de la membrana y se introduce el ADN.

15
Yo esperaría que las bacterias q se recombinaron sean las que crezcan en el lugar.

17
17

18
Enzimas de Restricción
1970’s endonucleasa de restricción bacterianas (enzimas de restricción) Reconocen secuencias específicas de la doble banda del ADN y lo cortan cerca o en el sitio de reconocimiento. 18
 Reconocen secuencias específicas de la doble banda del ADN y lo cortan cerca o en el sitio de reconocimiento. 18.")
19
Enzimas de restricción
Secuencias de pb Secuencias palindrómicas 5’-GAATTC-3’ 3’-CTTAAG-5’ Extremos pegagosos o romos La mayoría de las enzimas de restricción reconocen unos sitios de 4 o 6 pb, a pesar de que existen algunas que reconocen sitios de un mayor # de pb. Generalmente las secuencias son palindromicas: la secuencia de bases que reconocen se leen iguaL del extremo 5’ al 3’ en ambas hebras Extremos romos: cuando se cortan ambas hebras en un mismo punto, no tiene extremos pegajosos. 19

20
EcoRI Reconoce la secuencia específica de 6 pb
Corta en cada hebra entre las bases G y A Secuencias palindrómicas: 20

21
La enzima reconoce la región hace el corte
21

22
Luego hay otro número de enzimas que me permiten la unión, es con una ligasa que se da esta unión.
22

23
Otras enzimas de restricción reconocen diferentes secuencias de nucleótidos, secuencias especeificas para cada enzima Más de unas 1000 de tales enzimas se conocen y utilizan actualemnete Esta es una lista de las más usadas 23

24
Sitio de Reconocimiento Resultado del Corte EcoRI 5'GAATTC 3'CTTAAG
Enzima Origen Bacteriano Sitio de Reconocimiento Resultado del Corte EcoRI Escherichia coli 5'GAATTC 3'CTTAAG 5'---G**AATTC---3' 3'---CTTAA**G---5' BamHI Bacillus amyloliquefaciens 5'GGATCC 3'CCTAGG 5'---G**GATCC---3' 3'---CCTAG**G---5' HindIII Haemophilus influenzae 5'AAGCTT 3'TTCGAA 5'---A**AGCTT---3' 3'---TTCGA**A---5' Sau3A Staphylococcus aureus 5'GATC 3'CTAG 5'---**GATC---3' 3'---CTAG**---3' PovII* Proteus vulgaris 5'CAGCTG 3'GTCGAC 5'---CAG**CTG---3' 3'---GTC**GAC---5' Hay extremos pegajosos en la E. coli En la PovII no me deja extremos pegajosos por lo cual no me sirve para clonar.

25
colección reproducible y característica de fragmentos de ADN
La digestión de una molécula de ADN con una enzima de restricción determinada colección reproducible y característica de fragmentos de ADN refleja la frecuencia y localización de sitios específicos de restricción 25

26
Implicaciones 1- Un solo cambio de una sola base puede abolir el reconocimiento de la enzima por el sitio y por lo tanto su capacidad de corte en ese determinado lugar 2- Toda las moléculas de ADN digeridas con EcoRI, tienen los mismos extremos pegagosos. Esta propiedad de las enzimas de restricción tiene 2 implicaciones importantes centrales en su papel en la tecnología del AND recombinante y su aplicación a la genética médica. 1- La digestión de muestras de ADN genómico con, por ejemplo EcoRI, genera una colección de aproximadamente 1 millón de fragmentos, cada uno de una localización particular en el genoma. Como EcoRI corta el AND db especificamente cada vez que encuentra 5’-GAATTC-3’ y como un solo cambio de una sola base puede abolir el reconocimiento de la enzima por el; sitio y por lo tanto su capacidad ded corte en ese determinado lugar, tal digestión permite examinar esta secuancia de 6 pb en aprox 1 milloón de lugares en el genoma 2- Toda las moléculas de AND digeridas con EcoRi, tienen los mismos extremos pegagosos, independientemente de su origen y de la secuencias que se encuentren a los lados de las secuencias reconocidas por la enzima. Por lo tanto, dos moléculas cualquieras generadas por digestión con EcoRI pueden unirse in vitro por interacción con los overhangs complementarios de 4 pb, lo cual se logra por completion of the fosfodiester backbones on each strand por una enzima conocida como la ADN ligasa. Esta etapa de ligación crea una molécula de ADN recombinante, con una parte derivada de un ADN de una fuente y otra de otra fuente de ADN. 26

27
Foto de una electroforesis, el objetivo de esta es una migración de una molécula dependiendo de algunas condiciones.

28
28

29
Vectores “Molécula de ADN que se puede replicar de forma autónoma en un huesped (célula bacteriana o levadura) del cual puede ser aislado de una forma pura para su subsecuente análisis.” 29
 del cual puede ser aislado de una forma pura para su subsecuente análisis. 29.")
30
Los vectores pueden alcanzar un alto número de copias por célula.
Las bacterias y levaduras se pueden hacer crecer indefinidamente en un laboratorio grandes cantidades del ADN de interés El clonaje de fragmentos humanos de ADN en un vector por medio de enzimas de restricción y AND ligasa, permite la propagación del fragmentos de ADN clonado junto con la molécula del vector. El clonaje de fragmentos humanos de ADN en un vector por medio de enzimas de restricción y AND ligasa, permite la propagación del fragmentos de ADN clonado junto con la molécula del vector. 30

31
ADN recombinante “Nueva combinación de ADN creada in vitro entre secuencias de interés de ADN humano (u otro) y moléculas vectoras bacterianas (u otras) capaces de duplicarse indefinidamente dentro de una cepa de laboratorio” El nombre de ADN recombinante se refiere a la nueva combinación de AND creada in vitro entre secuencias de interés de AND humano (u otro) y moléculas vectoras bacterianas (u otras) capaces de duplicarse indefinidamente dentro de una cepa de laboratorio 31
 y moléculas vectoras bacterianas (u otras) capaces de duplicarse indefinidamente dentro de una cepa de laboratorio El nombre de ADN recombinante se refiere a la nueva combinación de AND creada in vitro entre secuencias de interés de AND humano (u otro) y moléculas vectoras bacterianas (u otras) capaces de duplicarse indefinidamente dentro de una cepa de laboratorio. 31.")
32
Vectores Plásmidos Bacteriófago Lambda Cósmidos BACs y YACs 32

33
Plásmidos ADN circular doble banda Se replica extracromosomalmente
En bacterias o levaduras Resistencia a antibióticos Segmentos cortos de ADN (hasta 15 kb) Selección pocos kb marcadores (resistencia a antibióticos) sitios de restricción Primero se descrubrieron en bacterias, ya que acarrean genes para la ressitencia a antiB y se pueden pasar facilmente de bacteria a bacteria Hasta 15 pares de bases se puede introducir en el plásmido para clonar. 33
 Selección. pocos kb. marcadores (resistencia a antibióticos) sitios de restricción. Primero se descrubrieron en bacterias, ya que acarrean genes para la ressitencia a antiB y se pueden pasar facilmente de bacteria a bacteria. Hasta 15 pares de bases se puede introducir en el plásmido para clonar. 33.")
34
Bacteriófago Lambda Virus bacterial ADN db relativamente largo (45kb)
1/3 de su genoma es no esencial Infecta las bacterias y se replica en la bacteria produciendo cantidades enormes de virus (hasta 1 millón) Puede crear lisis bacteriana Segmentos de ADN de hasta 20 kb 34
 Puede crear lisis bacteriana. Segmentos de ADN de hasta 20 kb. 34.")
35
Ciclo del bacteriografo
35

36
Cósmidos Plásmidos que utilizan la habilidad de las partículas infecciosas del bacteriófago lambda para empaquetar eficientemente piezas lineares de ADN y luego adherirse a la superficie de las bacterias e inyectar el contenido de ADN. Permite segmentos insertos de hasta 45 kb Secuencias Cos Es una seccion circular de ADN y el mecanismo de inserción del ADN es igual. 36

37
BACs Bacterial Artificial Chromosomes Plásmidos enormes 1990’s
Pueden contener insertos de hasta 300 kb 37

39
YACs Yeast Artificial Chromosomes
Vector con mayor capacidad (hasta 2000 kb) Se transfiere a Saccharomyces cerevisiae Se transfiere a Saccharomyces cerevisiae donde se replica y segrega como un cromosoma linear normal de levadura. 39
 Se transfiere a Saccharomyces cerevisiae. Se transfiere a Saccharomyces cerevisiae donde se replica y segrega como un cromosoma linear normal de levadura. 39.")
40
Se parte inicialmente de secuencias, el BAM HI es una enzima de restricción que general cortes homogéneos. Cuando se produce un cromosoma artificial hay que pensar en la estabilidad del cromosoma.

41
41

42
Librerías Genómicas 1- Digestión parcial del ADN genómico con una enzima de restricción. 2- Digestión del vector (misma ER) 3- Ligación del vector (ADN ligasa) 4- Introducción en célula huesped 42
 4- Introducción en célula huesped. 42.")
43
Librerías de ADN complementario
Ventajas: Representación directa de las secuencias codificantes (no intrones) Tejidos con expresión selectiva ADNc: copias de la población de ARNm presente en un determinado tejido. Transcriptasa reversa Problema: solo contiene los exones de un gen y no los intrones ni las secuencias promotoras. ==> no provee indicaciobnes acerca de la talla o el número de exones o acerca de la secuencia digerida. 43
 Tejidos con expresión selectiva. ADNc: copias de la población de ARNm presente en un determinado tejido. Transcriptasa reversa. Problema: solo contiene los exones de un gen y no los intrones ni las secuencias promotoras. ==> no provee indicaciobnes acerca de la talla o el número de exones o acerca de la secuencia digerida. 43.")
44
Transcriptasa Reversa
ADN polimerasa dependiente de ARN Derivada de retrovirus Requiere de primer para iniciar síntesis de ADN (oligo -dT). Oligo dT se une a la cola de poli A de los ARNm Niveles de ARNm transcripto Oligo dT se une a la cola de poli A de los ARNm PCR: se encuentra una región de interés y se amplifica 44
. Oligo dT se une a la cola de poli A de los ARNm. Niveles de ARNm transcripto. Oligo dT se une a la cola de poli A de los ARNm. PCR: se encuentra una región de interés y se amplifica. 44.")
45
45

46
Algunos vectores manufacturados contienen vectores de expresión: señles de transcripción o traducción adjacenjes al sitio de inserción del ADNc para facilitar la expresieon de la proteeina codificda por tal secuencia clonada. 46

47
Sondas de Acido Nucleico
identificación del clon que lleva la secuencia de interés screening de una librería hibridización del ácido nucleico Una vez que se realiza una librereia, la segunda etapa es la identificación del clon que lleva la secuencia de interés 47

48
Marcadores (probes) A una secuencia X de ácidos nucleicos se prueba su complementaridad con un fragmento de ADN o ARN conocido y marcado (que se puede detectar). Marcaje: Trazadores radioactivos (Fósforo 32) Compuestos histoquímicos Colorante fluorescente La especificidad de la hibridización del ácido nucleico con bandas complementarias hace posible el uso de probes Ej: fosforo 32 que dad su altaa energía puede exoponerse a placas de rayos X Se introduce el P32 a la secuencia conocida (marcadora) sustituyendo el P del enlace fosfodiester por P32 Si la secuencia X es complementaria se va a formar una doble hebra, que se puede detectar por la resencia del marcador => Se facilita la detección y aislamiento de la secuencia de interés. 48
. Marcaje: Trazadores radioactivos (Fósforo 32) Compuestos histoquímicos. Colorante fluorescente. La especificidad de la hibridización del ácido nucleico con bandas complementarias hace posible el uso de probes. Ej: fosforo 32 que dad su altaa energía puede exoponerse a placas de rayos X. Se introduce el P32 a la secuencia conocida (marcadora) sustituyendo el P del enlace fosfodiester por P32. Si la secuencia X es complementaria se va a formar una doble hebra, que se puede detectar por la resencia del marcador. => Se facilita la detección y aislamiento de la secuencia de interés. 48.")
49
acgacttgag 49

50
Hibridación Hebra simple desnaturalización
Formación de dobles hebras hibridización a fragmentos de ADN marcados con secuencia nucleótidica equivalente Desnaturalizacieon: por altas temperaturas alto pH bajas condiciones de sal 50

51
II- Reacción en Cadena de la Polimerasa
51

52
PCR Alternativa al clonaje
Generar cantidades ilimitadas de una secuencia de interés Es una amplificación enzimática de un fragmento de ADN (blanco o molde) situado entre dos oligonucleótidos “primers” PCR puede amplificar selectivamente una molécula individual de AND O ARN VARIOS BILLONES DE VECES en unas pocas horas. Ha revolucionado el diagneostico y análisis moleculares Se necesita especificidad si no se conoce la región no se puede hacer una PCR 52
 situado entre dos oligonucleótidos primers PCR puede amplificar selectivamente una molécula individual de AND O ARN VARIOS BILLONES DE VECES en unas pocas horas. Ha revolucionado el diagneostico y análisis moleculares. Se necesita especificidad si no se conoce la región no se puede hacer una PCR. 52.")
53
PCR ADN Primers: le dan la especificidad ADN polimerasa
Cofactores enzimáticos A,G,T,C

54
En el tubo está toda la cantidad del genoma
El ADN genómico que se extrae se ve abajo La actividad enzimática varía por: pH, temperatura, cofactores, concentración

55
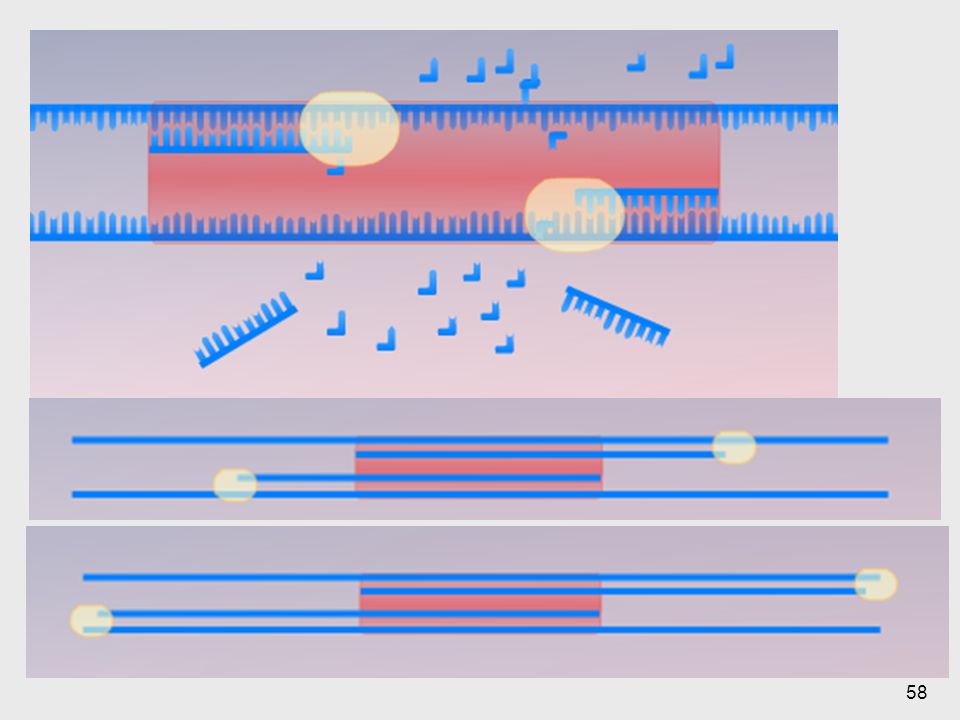
Se debe de hacer una desnaturalización (separar las dos bandas, romper puentes de hidrógeno, se rompe la estructura secundaria y terciara) y me libera cada uno de las cadenas
 y me libera cada uno de las cadenas")
56
Se logra que los primers encuentren las regiones, esto por medio de puentes de hidrógeno.

57
En esta se ve la temperatura a la cual la enzima va a poder polimerizar. El 3’ me da la región donde inicia.

59
Ciclo: se necesita los tres tiempo de temp: temp de desnaturalización, …. y polimerización

60
En el tercer ciclo se repite lo mismo y ahora tenemos una doble del tamaño que queríamos. Normalmente se hacen de ciclos.

61
Amplificación exponencial
2n n= # ciclos

62
62

63
Utilidad de la PCR Amplificar una región.(los primers se fabrican para servir en lugares donde no haya mutaciones. Algunos si lo hacen de esta manera podemos buscar mutaciones 63

64
PCR reverso Amplificación a partir de ARN ARNm ADNc amplificación
transcriptasa polimerasa reversa primers… Apartir del ARNm mensajero podemos obtener ADNc y luego se amplifica, lo que estariamos obteniendo serían exones sin intrones. Sirven para localizar la expresión de genes malignos como el cromosoma filadelfia. 64

65
diagnóstico por amplificación diagnóstico por ASO en SB
Porciones particulares del gen (usualmente exones) se pueden amplificar utilizando primers específicos para el gen normal o el gen mutado. diagnóstico por amplificación diagnóstico por ASO en SB 65
 se pueden amplificar utilizando primers específicos para el gen normal o el gen mutado. diagnóstico por amplificación. diagnóstico por ASO en SB. 65.")
66
66

67
Ventajas del PCR Requiere poca muestra Rápida
Barata (inversión inicial!!!) Sensibilidad y especificidad 67
 Sensibilidad y especificidad. 67.")
68
III- Métodos para el análisis de ácidos nucleicos
68

69
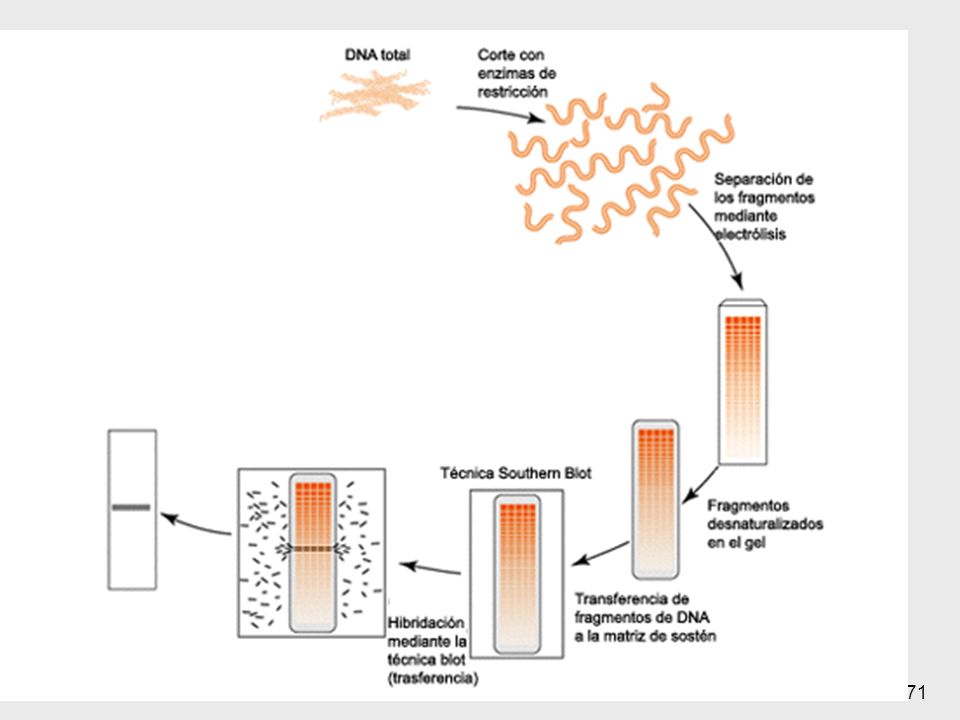
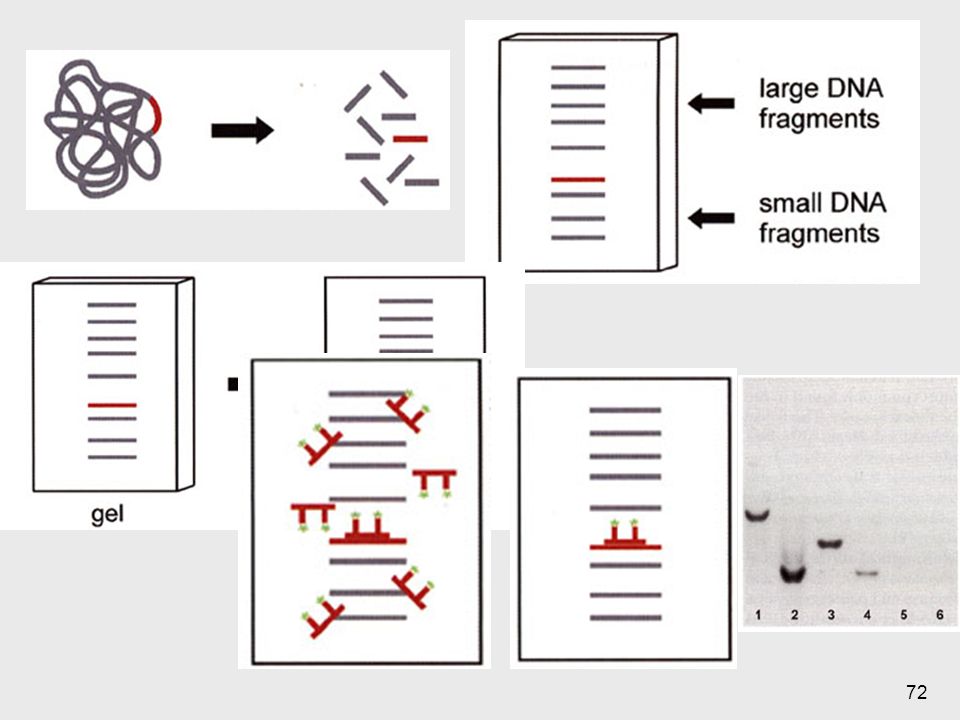
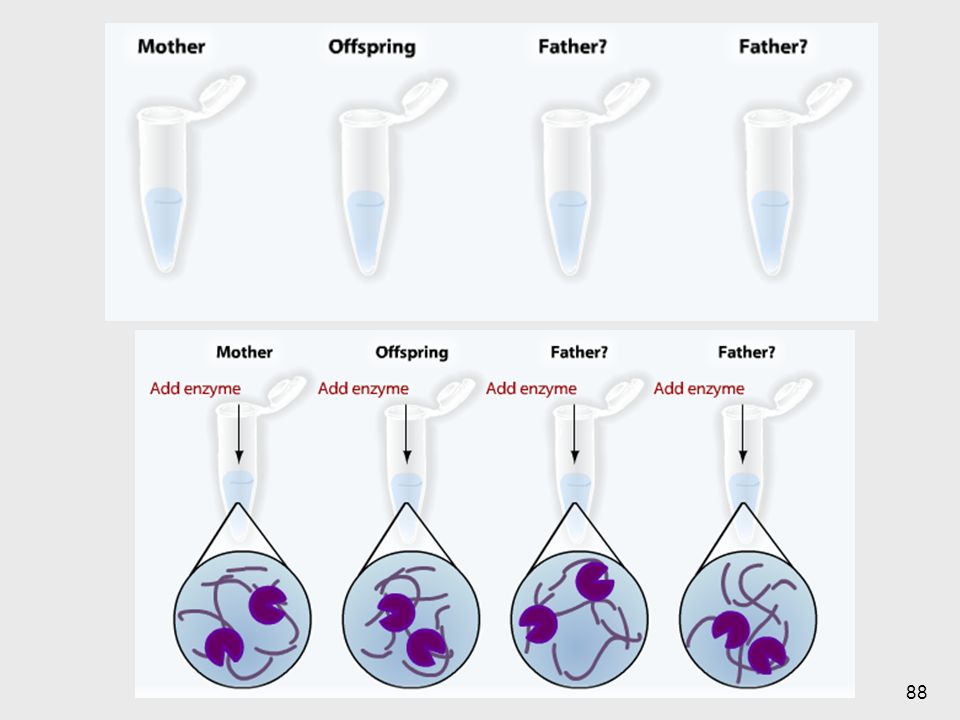
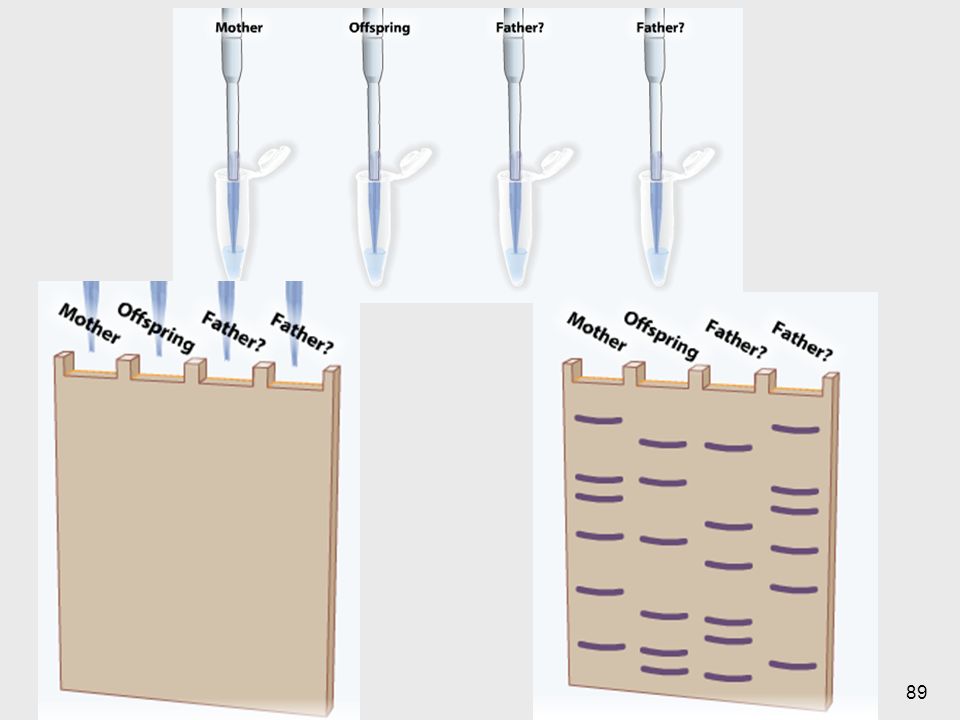
Southern Blotting Mediados de los 70’s
Método estándar para analizar la estructura del ADN digerido por enzimas de restricción 1- Aislamiento del ADN 10 ml de sangre => 108 leucocitos => 100µg de ADN 2- Digestión => ≈ 1 millón de fragmentos 3- Separación de los fragmentos según su tamaño ==> electroforesis en geles de agarosa 1- Cualquier célula se puede usar salvo los GR maduros, que no tienen núcleo. Generalmente se usan linfocitos obtenidos de sangre periférica Suficiente para docenas de reacciones de restricción También se pueden usar otros tejidos: fibroblastos cultivados de piel, líquido amniótico, vellocidades coriales, muestras de biopsias 69

70
5- Unión con una sonda marcada 6- Lavados
4- Blotting: A) desnaturalización (base fuerte) B) transferencia a un filtro de nitrocelulosa (blotting), por corriente eléctrica 5- Unión con una sonda marcada ==> hidridación 6- Lavados 7- Revelado (exposición al revelador, placa de rayos X…) 70
 desnaturalización (base fuerte) B) transferencia a un filtro de nitrocelulosa (blotting), por corriente eléctrica. 5- Unión con una sonda marcada. ==> hidridación. 6- Lavados. 7- Revelado (exposición al revelador, placa de rayos X…) 70.")
73
73

74
Electroforesis en geles de agarosa
Separa las moléculas por tamaño Fragmentos pequeños migran más rapidamente que los grandes Tinción con bromuro de etidio ==> Fluoresce con luz UV Bromuro de etidio: mutagénico y cancerígeno Fluoresce con luz UV Se ve como smear ya que hay una cantidad demasiado alta de fragmentos como para poder verlos individualmente. Bromuro de Etilo se pega al ADN y se ilumina con luz UV. Esta sustancia es mutagenica y cancerígena por tal motivo hay que manejarla con mucho cuidado. Usando electroforesis separaremos el ADN por su carga negativa. 74

75
NEGATIVO POSITIVO

76
La

77
Análisis con sondas alelo específicas
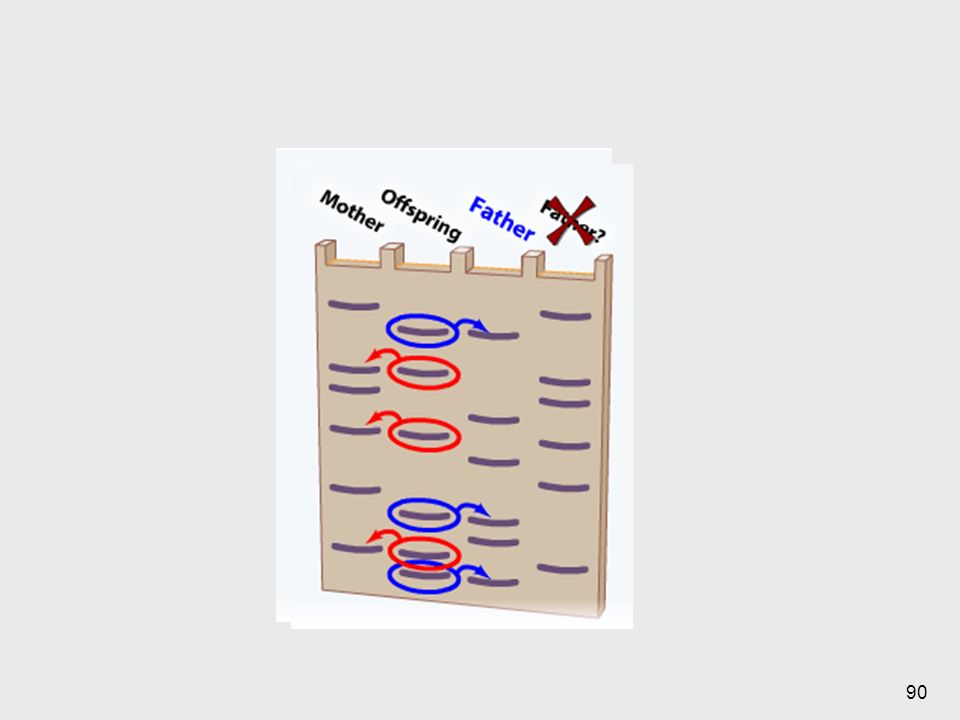
Sonda sintética de oligonucleótidos = ASO (Allele Specific Oligonucleotide) Corta longitud aprox. 20 pb ===> sensible a diferencias en un par de bases ASOs para el alelo normal o para el alelo mutado Permite diferenciar homocigotas y heterocigotas Cuando en una enfermedad geneetica se conoce la mutación en epsceifico que causa el caudro, , se puede analisar los diferentes pacientes en busqueda de si tienen o no la mutación. La mejor sonda para esto es una sonda sintéitica de oligonucleótidos Es importante reconocer la diferencia entre los análisi con ASOs y el análisis convencional de Southern Blott con sondas de AND clonado. Con el AND clonado no se puede revelar un cambio de 1 sola base (a menos de que este variación cree o destruya un sitio de restriccieon, alterando así el tamaño del fragmento detectado ppor la sonda). \En la mayoría de los casos, el cambio de 1 base o delceciones o inserciones pequeñas no se pueden distinguir por medio de un SB estándar, solamente se puede lograr utilizando ASOs. 77
 Corta longitud aprox. 20 pb ===> sensible a diferencias en un par de bases. ASOs para el alelo normal o para el alelo mutado. Permite diferenciar homocigotas y heterocigotas. Cuando en una enfermedad geneetica se conoce la mutación en epsceifico que causa el caudro, , se puede analisar los diferentes pacientes en busqueda de si tienen o no la mutación. La mejor sonda para esto es una sonda sintéitica de oligonucleótidos. Es importante reconocer la diferencia entre los análisi con ASOs y el análisis convencional de Southern Blott con sondas de AND clonado. Con el AND clonado no se puede revelar un cambio de 1 sola base (a menos de que este variación cree o destruya un sitio de restriccieon, alterando así el tamaño del fragmento detectado ppor la sonda). \En la mayoría de los casos, el cambio de 1 base o delceciones o inserciones pequeñas no se pueden distinguir por medio de un SB estándar, solamente se puede lograr utilizando ASOs. 77.")
78
diagnóstico por amplificación diagnóstico por ASO en SB
Porciones particulares del gen (usualmente exones) se pueden amplificar utilizando primers específicos para el gen normal o el gen mutado. diagnóstico por amplificación diagnóstico por ASO en SB 78
 se pueden amplificar utilizando primers específicos para el gen normal o el gen mutado. diagnóstico por amplificación. diagnóstico por ASO en SB. 78.")
79
Hay que tener cuaidado al interpretar los resultados. Con ASOs
==> no todos los genes mutantes de un detreminado locus comparten la misma alteración en la secuencia del AND. Por lo tanto el que no se hibridice con la sonda ASO para detectar el mutante en cuestión no garantiza que el gen del paciente es normal, ya que puede tener una mutación en otro lugar que no es la zona abarcada por ese ASO. Los análisis con ASOs se usan cuando hay una alta probabilidad (por lineas o comportamientos familiares) de que ese individuo posea esa mutación. Se pueden usar los primers para valorar la presencia de mutaciones pues cuando las mismas esten presentes entonces se van a pegar. Se utilizan primers de mutacion y de AND normal, entonces se aplican para ver si una persona posee genes mutantes, normales o ambos (hetero/homocigoto) y además del gen mutante tambien tiene un alelo normal. Thompson & Thompson Genetics in Medicine 79
 de que ese individuo posea esa mutación. Se pueden usar los primers para valorar la presencia de mutaciones pues cuando las mismas esten presentes entonces se van a pegar. Se utilizan primers de mutacion y de AND normal, entonces se aplican para ver si una persona posee genes mutantes, normales o ambos (hetero/homocigoto) y además del gen mutante tambien tiene un alelo normal. Thompson & Thompson Genetics in Medicine. 79.")
81
IV- Métodos para análisis de proteínas
81

82
Western Blotting 1- Aislamiento de las proteínas de la muestra
2- Separación por electroforesis en geles de poliacrilamida 3- Transferencia a membrana 4- Incubación de la membrana con Ac anti -proteína en cuestión 5- Adición de un segundo Ac marcado 6- Revelado Muchas veces se requiere saber no solamente cual es el defecto en la molécula de AND, sino también a que punto este defecto altera la proteína codificda involucrada en el cuadro clínico. Información acerca del tamaño y la cantidad de proteína mutante en células de pacientes con enfermedades genéticas Se aislan proteinas se separan mediante electroforesis y luego se evidencian estan proteínas que pueden ser antigenos como los de SIDA, mediante marcadores fosforecentes y se visualizan. 82

83
Se utiliza una sust que le de carga negativa a la proteína
Se utiliza una sust que le de carga negativa a la proteína. En un Duchenne clásico vemos como desaparece la proteína, en los becker simplemente disminuye de tamaño 83

84
V- Análisis de la secuencia de ADN
84

85
Secuenciación de Sanger
Análogos de nucleótidos que inhiben la polimerasa “Materiales”: ADN molde a secuenciar Primers ADN polimerasa dNTPs ddNTPs análogos marcados (fluorescencia o radiación) Electroforesis en gel de agarosa Fred Sanger, Premio Nobel en 1980 por tecnica de secuenciación del ADN Análogos de nucleótidos que inhiben la polimerasa a medida que sintetiza la banda complementaria al ADN molde que se está secuenciando. 85
 Electroforesis en gel de agarosa. Fred Sanger, Premio Nobel en 1980 por tecnica de secuenciación del ADN. Análogos de nucleótidos que inhiben la polimerasa a medida que sintetiza la banda complementaria al ADN molde que se está secuenciando. 85.")
86
86

87
LA secuacion 87

91
VI- Hibridación in situ de cromosomas
91

92
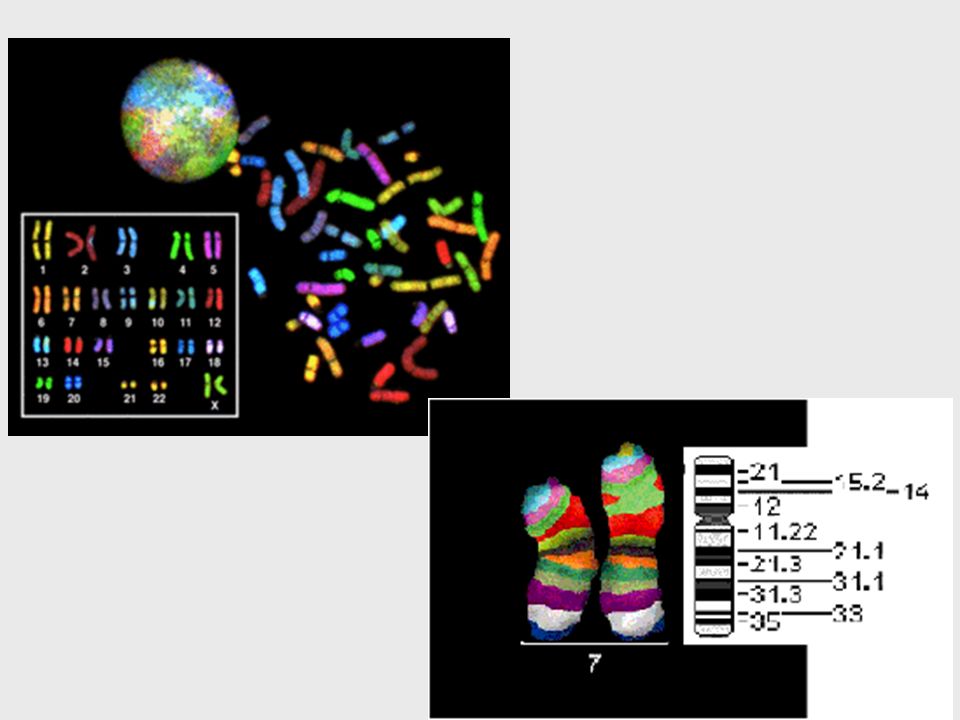
FISH (hibridación fluorescente in situ )
Cromosomas en metafase se fijan en portaobjetos Se desnaturalizan in situ ==> se exponen las dos hebras del ADN Se hibridizan con sondas marcadas con un colorante fluorescente Se visualizan al microscopio de fluorescencia con una longitud de onda que y un filtro para observar el fluorocromo 92

94
“Chromosome painting”
Parte de un brazo cromosómico Todo un brazo El cromosoma entero Spectral karyotiping (SKY) Análisis de telómeros / microdeleciones 94
 Análisis de telómeros / microdeleciones. 94.")
95
Mapeo por FISH Permite visualización directa de la posición del gen
Regiones de 1-2 millones de pb (cromatina condensada) Los anteriores son métodos indirectos, no se puede ver la posición del gen directamente 95
 Los anteriores son métodos indirectos, no se puede ver la posición del gen directamente. 95.")
98
Un caso interesante de transferencia de genes se ilustra en este experimento en el que aisló el gen para la enzima luciferasa de las luciérnagas y se incorporó en esta planta. El sustrato para la enzima luciferasa es una proteína llamada luciferina. En presencia de oxígeno, la luciferina junto con luciferasa y ATP producen bioluminiscencia, como se ve en el destello de la luciérnaga. El gen de la luciferasa fue clonado en E. coli y luego empalmado en el cromosoma de un virus vegetal, lo cual le suministró una secuencia regulatoria. El cromosoma viral modificado se insertó luego en plásmidos Ti, los plásmidos fueron transferidos a las bacterias y las bacterias se incubaron con células foliares del tabaco. Las células formaron una masa de tejido, conocido como callo, a partir del cual se obtuvieron nuevas plantas en medio de crecimiento adecuado. Las nuevas plantas fueron regadas con una solución que contenía luciferina y al cabo de un tiempo las plantas resplandecían. 98

99
Literatura: Nelson D, Cox M. Lehninger Principles in Biochemistry. Strachan T, Read A. Human Molecular Genetics Nussbaum R, McInnes R, Willard H. Thompson & Thompson Genetics in Medicine. Bases de datos:

Presentaciones similares












 Noviembre de 2004.>")









>")
