Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Muerte súbita: prevención primaria
Óscar Guzmán Ruiz Mayo 2011

2
Escenario clínico y búsqueda
Paciente: Mujer de 47 años sin antecedentes de interés que sobrevive a parada cardíaca Intervención: realización de cribado en familiares Resultado: 1.- Rentabilidad del estudio en familiares 2.- Intervención en resultados positivos 3.- Identificación del papel de los estudios genéticos Términos de búsqueda: sudden death, cardiac death, cardiac arrest, familial screening, genetic testing Sitios de búsqueda (5S): UpToDate, Tripdatabase, Pubmed, Medicine, Revista española de Cardiología, Guideline.gov, Google
: UpToDate, Tripdatabase, Pubmed, Medicine, Revista española de Cardiología, Guideline.gov, Google.")
3
Parada súbita cardíaca (PSC)
“Es el cese súbito de actividad cardíaca dejando a la víctima arreactiva, sin respiración normal y sin signos de circulación. Si no se toman medidas correctoras rápidamente, esta condición progresa a muerte súbita.” (Documento de la ACC/AHA/HRS de 2006) muertes en EEUU entre 1998 y 1999. 63% de las muertes cardíacas en ≥ 35 años
 muertes en EEUU entre 1998 y % de las muertes cardíacas en ≥ 35 años.")
4
Causas de PCS Cardiopatía isquémica coronaria: 65-70%
Otros tipos de enfermedad cardíaca estructural: 10% Corazón estructuralmente normal: 5-10% Causas NO CARDIACAS: 15-35% Insuficiencia cardíaca (30-50% muertes en IC) REVERSIBLES: Isquemia aguda Fármacos antiarrítmicos Medicamentos (prolongadores QT) Alteraciones electrolíticas: hipopotasemia, hiperpotasemia, hipomagnesemia I. Cardíaca
 REVERSIBLES: Isquemia aguda. Fármacos antiarrítmicos. Medicamentos (prolongadores QT) Alteraciones electrolíticas: hipopotasemia, hiperpotasemia, hipomagnesemia. I. Cardíaca.")
5
ENFERMEDAD CARDÍACA NO ISQUÉMICA NO PATOLOGÍA CARDÍACA
ISQUEMIA CARDÍACA Enfermedad eléctrica primaria (fibrilación ventricular idiopática) Enfermedad coronaria con infarto miocárdico o angor Sd. Brugada Embolia de arteria coronaria Sd. QT largo y QT corto Patología coronaria no aterogénica (arteritis, diseccion, anomalías congénitas) Sd. De preexcitación (Wolff- Parkinson-White) Bloqueo completo cardíaco Espasmo coronario Muerte súbita familiar ENFERMEDAD CARDÍACA NO ISQUÉMICA Trauma de pared torácica (conmotio cordis) NO PATOLOGÍA CARDÍACA Cardiopatía hipertrófica Cardiopatía dilatada TEP Valvulopatía (incl. prolapso mitral) Hemorragia intracraneal Enfermedad cardíaca congénita Ahogamiento Displasia arritmogénica de ventrículo derecho (ARVD) Drogas-Fármacos Obstrucción de vía respiratoria central Miocarditis Taponamiento cardíaco agudo Síndrome de muerte súbita infantil Ruptura miocardica aguda Disección aórtica SIN PATOLOGÍA CARDÍACA ESTRUCTURAL
 Enfermedad coronaria con infarto miocárdico o angor. Sd. Brugada. Embolia de arteria coronaria. Sd. QT largo y QT corto. Patología coronaria no aterogénica (arteritis, diseccion, anomalías congénitas) Sd. De preexcitación (Wolff- Parkinson-White) Bloqueo completo cardíaco. Espasmo coronario. Muerte súbita familiar. ENFERMEDAD CARDÍACA NO ISQUÉMICA. Trauma de pared torácica (conmotio cordis) NO PATOLOGÍA CARDÍACA. Cardiopatía hipertrófica. Cardiopatía dilatada. TEP. Valvulopatía (incl. prolapso mitral) Hemorragia intracraneal. Enfermedad cardíaca congénita. Ahogamiento. Displasia arritmogénica de ventrículo derecho (ARVD) Drogas-Fármacos. Obstrucción de vía respiratoria central. Miocarditis. Taponamiento cardíaco agudo. Síndrome de muerte súbita infantil. Ruptura miocardica aguda. Disección aórtica. SIN PATOLOGÍA CARDÍACA ESTRUCTURAL.")
6
Evaluación general del superviviente a PSC
Identificación y tratamiento de causas reversibles Valoración de enfermedad cardíaca estructural: Historia, exploración, analítica, EKG, Ecocardiograma, coronariografía (1997, Joint Steering Committee of the Unexplained Cardiac Arrest Registry) En pacientes SIN desencadenantes arritmogénicos obvios o anomalías cardíacas estructurales, realización de estudios de enfermedades eléctricas primarias Valoración neurológica y psicológica En pacientes seleccionados con sospecha/confirmación de enfermedades heredables: valoración de familiares
 En pacientes SIN desencadenantes arritmogénicos obvios o anomalías cardíacas estructurales, realización de estudios de enfermedades eléctricas primarias. Valoración neurológica y psicológica. En pacientes seleccionados con sospecha/confirmación de enfermedades heredables: valoración de familiares.")
7
Valoración de Enfermedades eléctricas primarias
Canalopatías: Sd. Brugada Sd. QT largo Sd. QT corto Taquicardia ventricular polimórfica catecolaminérgica (CPVT) No canalopatía Sd. Wolff-Parkinson-White Conmotio cordis ESTUDIOS Estudio electrofisiológico Ergometría Holter Test provocación farmacológica: adrenalina, procainamida, flecainida…
 No canalopatía. Sd. Wolff-Parkinson-White. Conmotio cordis. ESTUDIOS. Estudio electrofisiológico. Ergometría. Holter. Test provocación farmacológica: adrenalina, procainamida, flecainida…")
8
El 18% de las MS ocurrieron durante el ejercicio físico, el 32% en actividades cotidianas y el 37% en sueño/reposo

9
Miocardiopatía hipertrófica
Excesiva hipertrofia del ventrículo izquierdo, correlacionada con disfunción diastólica. Volumen de VI normal Herencia predominantemente A.D. Es la causa más frecuente en EEUU de MS en sujetos jóvenes (<35 años) y en atletas de competición. Factores de riesgo: síncopes inexplicados, Hª Familiar de MS, septo en diástole >3 cm, una o más rachas de TV a más de 120 spm, ausencia de elevación de PA en más de mmHg en ergometría 2 o más factores de riesgo predicen tasa de MS del 3-6%
 y en atletas de competición. Factores de riesgo: síncopes inexplicados, Hª Familiar de MS, septo en diástole >3 cm, una o más rachas de TV a más de 120 spm, ausencia de elevación de PA en más de mmHg en ergometría. 2 o más factores de riesgo predicen tasa de MS del 3-6%")
10
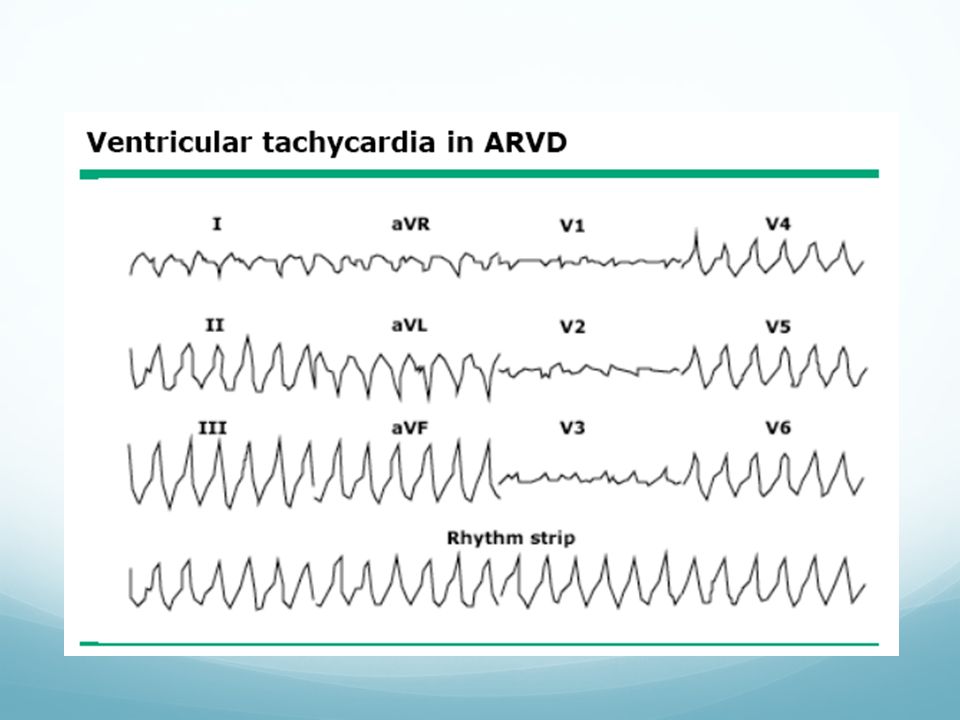
Displasia arritmogénica de ventrículo derecho (ARVC)
Progresiva infiltración grasa en ventrículo derecho, asociado a arritmias ventriculares Prevalencia 1:1000. En Italia más Clínica: palpitaciones (27%), síncope (26%), MS (23%). Entre 2ª y 5ª década de la vida Característica morfología de BRI con eje inferior Diagnóstico por RNM cardíaca Tratamiento: antiarrítmicos, ablación y DAI
, síncope (26%), MS (23%). Entre 2ª y 5ª década de la vida. Característica morfología de BRI con eje inferior. Diagnóstico por RNM cardíaca. Tratamiento: antiarrítmicos, ablación y DAI.")
11
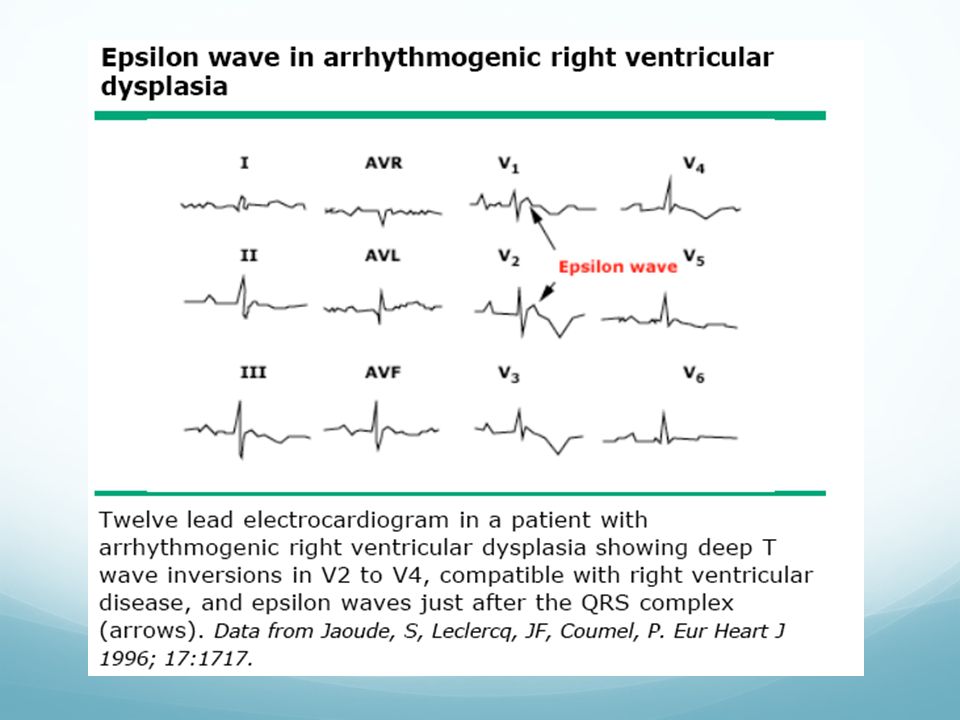
Criterios diagnósticos de ARVD
Historia familiar: enfermedad familiar confirmada con necropsia, cirugía. Historia familiar de muerte súbita prematura (<35 años), causada por sospecha de ARVD EKG: anormalidad de despolarización/conducción: ondas EPSILON o prolongación localizada (>110 ms) del complejo QRS en derivaciones precordiales derecha (V1-3) EKG: anormalidades de repolarización: T invertida en derivaciones precordiales derechas (V2-3) en individuos de más de 12 años de edaden ausencia de BRDHH Arritmias: taquicardias ventriculares tipo BRI sustenidas o no documentadas en EKG, Holter o ergometría. Extrasistolia ventricular frecuente (>1000 en 24h) Disfunción global o regional y alteraciones estructurales: dilatación grave y reducción de la FE de VD sin afectación de VI, aneurismas ventriculares derechas, dilatación segmentaria grave del VD Histología: recambio fibrograso de miocardio en biopsia (Tomado de: Corrado D et al. Arritmogenic right ventricular cardiomiopathy: diagnosis, prognosis and treatment. Heart. 2000;83: )
, causada por sospecha de ARVD. EKG: anormalidad de despolarización/conducción: ondas EPSILON o prolongación localizada (>110 ms) del complejo QRS en derivaciones precordiales derecha (V1-3) EKG: anormalidades de repolarización: T invertida en derivaciones precordiales derechas (V2-3) en individuos de más de 12 años de edaden ausencia de BRDHH. Arritmias: taquicardias ventriculares tipo BRI sustenidas o no documentadas en EKG, Holter o ergometría. Extrasistolia ventricular frecuente (>1000 en 24h) Disfunción global o regional y alteraciones estructurales: dilatación grave y reducción de la FE de VD sin afectación de VI, aneurismas ventriculares derechas, dilatación segmentaria grave del VD. Histología: recambio fibrograso de miocardio en biopsia (Tomado de: Corrado D et al. Arritmogenic right ventricular cardiomiopathy: diagnosis, prognosis and treatment. Heart. 2000;83: )")
14
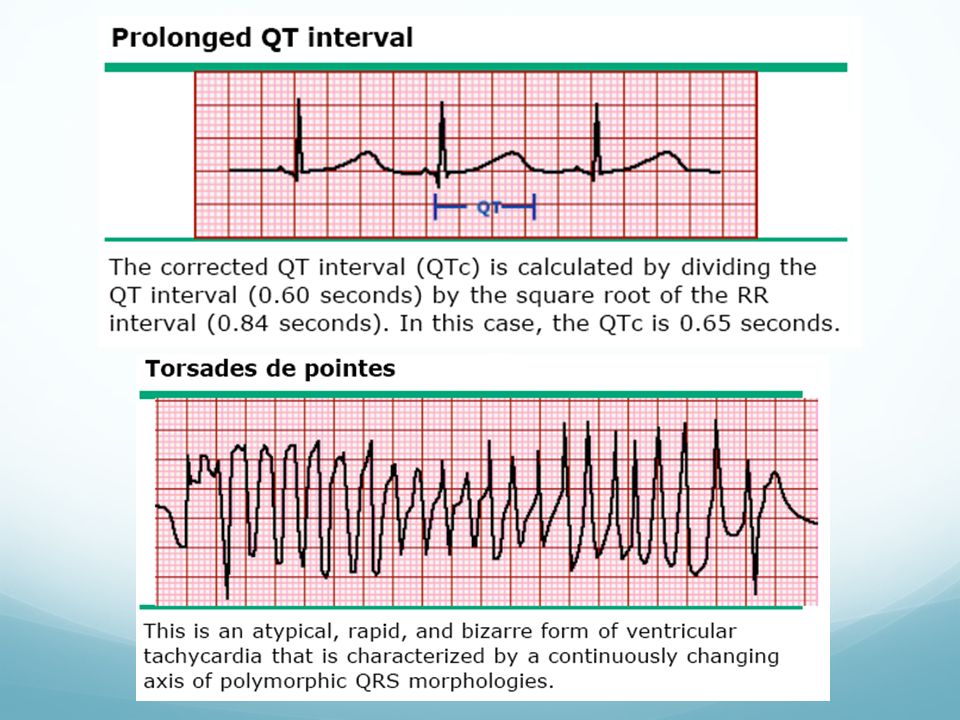
Síndrome de QT Largo Trastorno hereditario (A.D. o A.rec.) o adquirido. Anormal prolongación del intervalo QT y asocia arritmias ventriculares: Torsade de Pointes Prevalencia 1:10.000 En las formas congénitas la primera manifestación suele ser en la niñez o adolescencia (5 a 15 años) Formas adquiridas: quinidina, procainamida, sotalol, amiodarona, ADT, dietas protéicas e inecticidas Pronóstico: grado de prolongacion de QT y existencia previa de parada cardíaca Tratamiento: reducción de actividad física, evitar fármacos implicados. Uso de betabloqueantes y DAI (supervivientes de MS)
 Formas adquiridas: quinidina, procainamida, sotalol, amiodarona, ADT, dietas protéicas e inecticidas. Pronóstico: grado de prolongacion de QT y existencia previa de parada cardíaca. Tratamiento: reducción de actividad física, evitar fármacos implicados. Uso de betabloqueantes y DAI (supervivientes de MS)")
15
Criterios diagnósticos SQTL hereditario
EKG: Prolongación de QT; torsad de pointes, alternancia de onda T, morfología de onda T Clínica: síncope, sordera congénita Hª Familiar: familiares con SQTL establecido, MS con <30 años entre familiares inmediatos (Tomado de Schwartz PJ. Diagnostic criteria for the long QT syndrome: an update. Circulation. 1993;88: )
")
19
Síndrome QT Corto Descrito en 2000. QTc ≤340 ms
Alto riesgo de síncope y arritmias ventriculares malignas. FA en edad joven. Niños y jóvenes sin cardiopatía estructural Hiperfunción canales de K: Genes KCNH2 (=HERG), KCNQ1 y KCNJ2
, KCNQ1 y KCNJ2.")
20
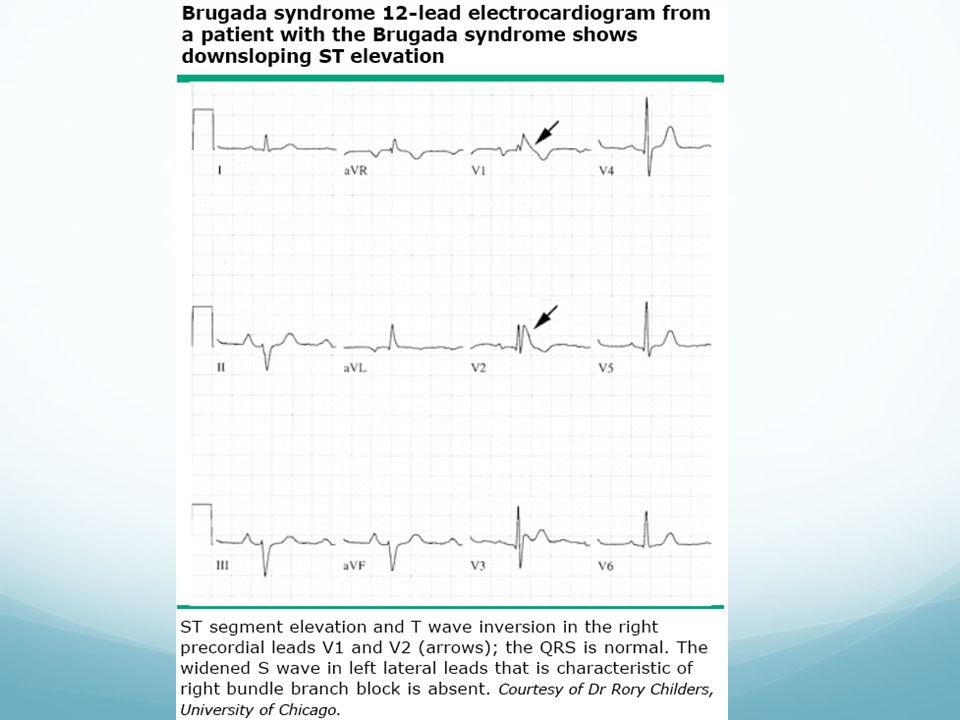
Síndrome de Brugada Descrito en 1992. Asocia MS
BRD, elevación persistente de ST en precordiales derechas Tipo1: elevación en cubeta del segmento ST ≥ 2mm seguida de una onda T negativa, con ausencia de separación isoeléctrica, presente en más de una derivación derecha (V1-3) Tipo2: Elevación de ST seguida de T positiva trifásica que adquiere morfología de “silla de montar” Tipo3: Elevación de ST ≤ 1 mm, con patrón de cubeta o silla de montar El patrón puede fluctuar en el tiempo, pudiendo ser normal transitoriamente (desenmascara: fiebre, activación vagal, fac. hormonales y fármacos) Diagnóstico: Tipo 1: patrón descrito en más de 3 derivacipnes junto a a.) FV documentada; b.) episodios de TV polimórfica; c.)inductibilidad de arritmias ventriculares en EEF; d.) Hª Familiar de MS <45 años; e.)presencia de patrón tipo I en algún miembro de la familia; f.) síncope y g.) respiración agónica nocturna. En el tipo 2 y 3: test de flecainida o ajmalina (provoca cambio a patrón tipo 1) Pronóstico peor: varón, síntomas previos al diagnóstico, patrón 1, inducibilidad de arritmias en EEF Tratamiento: Sintomáticos: DAI Asintomáticos: Positivo: DAI, Negativo: seguimiento Familiares: realizar EEF y DAI según resultado Genética: Gen SCN5A
 Tipo2: Elevación de ST seguida de T positiva trifásica que adquiere morfología de silla de montar Tipo3: Elevación de ST ≤ 1 mm, con patrón de cubeta o silla de montar. El patrón puede fluctuar en el tiempo, pudiendo ser normal transitoriamente (desenmascara: fiebre, activación vagal, fac. hormonales y fármacos) Diagnóstico: Tipo 1: patrón descrito en más de 3 derivacipnes junto a a.) FV documentada; b.) episodios de TV polimórfica; c.)inductibilidad de arritmias ventriculares en EEF; d.) Hª Familiar de MS <45 años; e.)presencia de patrón tipo I en algún miembro de la familia; f.) síncope y g.) respiración agónica nocturna. En el tipo 2 y 3: test de flecainida o ajmalina (provoca cambio a patrón tipo 1) Pronóstico peor: varón, síntomas previos al diagnóstico, patrón 1, inducibilidad de arritmias en EEF. Tratamiento: Sintomáticos: DAI. Asintomáticos: Positivo: DAI, Negativo: seguimiento. Familiares: realizar EEF y DAI según resultado. Genética: Gen SCN5A.")
23
Taquicardia ventricular polimórfica catecolaminérgica (CPVT)
Arritmias graves en pacientes jóvenes con corazón aparentemente normal y QT normal Mediadas por estrés o ejercicio. Diagnóstico: ergometría Clínica: síncopes o MS. Entre los 3-16 años Mortalidad sin tratamiento: 30-50% Tratamiento: dosis máxima de Betabloqueantes junto a implantación de DAI Base genética heterogénea

25
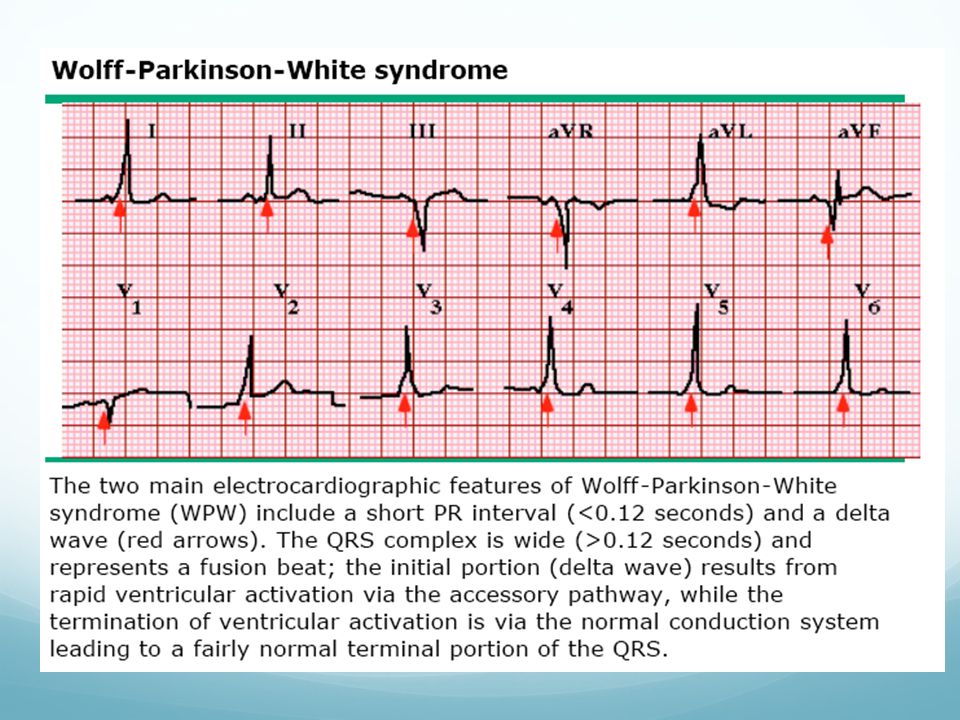
Síndrome de Wolff-Parkinson-White
Asociación de preexcitación ventricular con taquiarritmias auriculares. Asocia MS Incidencia entre 0,15-1% Factores de riesgo: Hª previa de taquiarritmias auriculares (FA!), que pueden degenerar en arritmias ventriculares Tratamiento: ablación con radiofrecuencia (93% éxito) en pacientes con arritmias sintomáticas
, que pueden degenerar en arritmias ventriculares. Tratamiento: ablación con radiofrecuencia (93% éxito) en pacientes con arritmias sintomáticas.")
27
Fibrilación ventricular idiopática
Diagnóstico de exclusión Afecta a varones jóvenes Excelente pronóstico con DAI

28
Problemas con los genes
Gran número de genes implicados Numerosas mutaciones para cada gen (RyR2) Expresión y penetración variable Costo elevado Significado incierto de la presencia de mutaciones de estos genes en asintomáticos Esto ocurre en muchas enf. Cardiovasculares, con la excepción de la enfermedad de Marfan Cribado genético en centros especializados arroja diagnósticos en un 33% de los jóvenes con MS sin cardiopatía estructural
 Expresión y penetración variable. Costo elevado. Significado incierto de la presencia de mutaciones de estos genes en asintomáticos. Esto ocurre en muchas enf. Cardiovasculares, con la excepción de la enfermedad de Marfan. Cribado genético en centros especializados arroja diagnósticos en un 33% de los jóvenes con MS sin cardiopatía estructural.")
30
Enfermedades geneticamente estudiables
Test genéticos en enfermedades con puntuación: ≥3: Miocardiopatía hipertrófica, Sd. QT largo, miocardiopatía dilatada con defectos de conducción y CPVT Entre 1 y 3 (dudoso): ARVD, Sd. Brugada ≤1 (no recomendado): miocardiopatía dilatada
: ARVD, Sd. Brugada. ≤1 (no recomendado): miocardiopatía dilatada.")
31
Genética de la MS Sd. Brugada: SCN5A, presente en 20-25%
Sd. QT corto: HERG, KCNQ1 y KCNJ2 ARVD: 5 genes desmosales (JUP, DSP, PKP-2, DSG-2 y DSG-2), RyR2 y TGF-β3 Miocardiopatía hipertrófica: cadena pesada de β-miosina y proteína C de unión a miosina. Es la enfermedad cardiovascular heredable más frecuente Sd. QT largo: A.Dom. (Romano-Ward): KCNQ1, KCNH2, SCN5A, ANKB A. Rec. (Large-Nielsen): KCNQ1, KCNE Sd. QT corto: CPVT: presente en 70%: A. Dom: RyR2 (muy largo) A- Rec CASQ2
, RyR2 y TGF-β3. Miocardiopatía hipertrófica: cadena pesada de β-miosina y proteína C de unión a miosina. Es la enfermedad cardiovascular heredable más frecuente. Sd. QT largo: A.Dom. (Romano-Ward): KCNQ1, KCNH2, SCN5A, ANKB. A. Rec. (Large-Nielsen): KCNQ1, KCNE. Sd. QT corto: CPVT: presente en 70%: A. Dom: RyR2 (muy largo) A- Rec CASQ2.")
32
Valoración de 32 familias de víctimas de MS inexplicada. Sujetos: 107
ECG, Ecocadio, Holter y ocasionalmente ergometría Siete familias (32%) fueron diagnosticadas de: Sd., QT largo: 4 Enfermedad cardíaca no estructural: 1 Distrofia miotónica: 1 Miocardiopatía hipertrófica: 1
 fueron diagnosticadas de: Sd., QT largo: 4. Enfermedad cardíaca no estructural: 1. Distrofia miotónica: 1. Miocardiopatía hipertrófica: 1.")
33
57 familias con AP de MS. ECG, Ecocardio (test ajmalina, RNM, test genéticos)
Resultados: 13 familias (30%) tenían AP de otras MS 30 familias (53%) fueron diagnosticadas de cardiopatía heredable: Sd. QT largo: 13, (+3 posibles) SD. Brugada: 5 ARVC: 5 Otras miocardiopatías: 4 Test genéticos: SCN5A (1) y KCNH2 (4) en una familia con Sd. QT largo (38%) SCN5A (1) en Sd. Brugada (20%) PKP2 (1) en ARVC (25%)
 tenían AP de otras MS. 30 familias (53%) fueron diagnosticadas de cardiopatía heredable: Sd. QT largo: 13, (+3 posibles) SD. Brugada: 5. ARVC: 5. Otras miocardiopatías: 4. Test genéticos: SCN5A (1) y KCNH2 (4) en una familia con Sd. QT largo (38%) SCN5A (1) en Sd. Brugada (20%) PKP2 (1) en ARVC (25%)")
35
43 familias afectas de MS con 150 miembros
Se identificó enfermedad heredable en 17 (40%) familias: Taquicardia ventricular polimórfica catecolaminérgica: 5 Sd. QT largo: 4 Sd. Brugada: 2 Sd. Brugada/QT largo: 1 Displasia arritmogénica de VD: 3 Miocardiopatía hipertrófica: 1 Hipercolesterolemia familiar: 1 Confirmación por estudios genéticos en 10 familias Portadores asintomáticos: media de 8,9 por familia Mayor probabilidad diagnóstica si: ≥ 2 episodios de MS inexplicados Más familiares investigados
 familias: Taquicardia ventricular polimórfica catecolaminérgica: 5. Sd. QT largo: 4. Sd. Brugada: 2. Sd. Brugada/QT largo: 1. Displasia arritmogénica de VD: 3. Miocardiopatía hipertrófica: 1. Hipercolesterolemia familiar: 1. Confirmación por estudios genéticos en 10 familias. Portadores asintomáticos: media de 8,9 por familia. Mayor probabilidad diagnóstica si: ≥ 2 episodios de MS inexplicados. Más familiares investigados.")
37
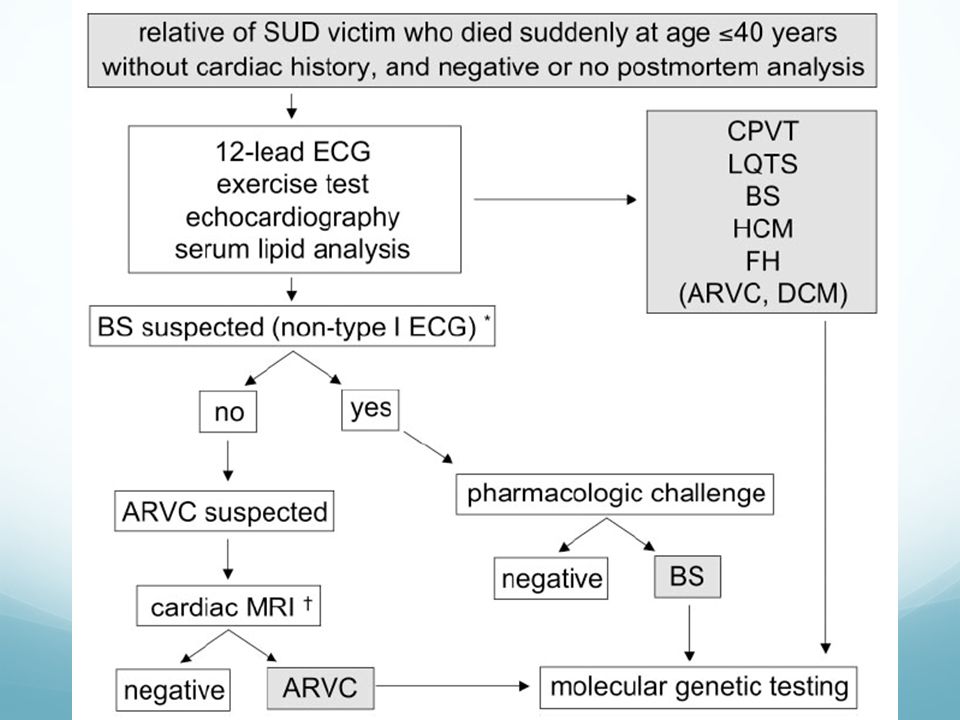
Algoritmo de Prevención Primaria en MS. MEDICINE

38
Algoritmo de prevención primaria de MS en cardiopatía por alteración eléctrica y población general.
MEDICINE

39
Valoración de familiares
Un episodio de MS confiere a familiares un riesgo entre 1,6 y 1,8 Procedimiento mínimo en familiares de PRIMER y SEGUNDO GRADO: anamnesis, exploración, analítica, ECG, ergometría, Holter 24h, Ecocardio Accesorio: coronariografía y ventriculografía, EEF, RNM EEF: las TV polimorficas ventriculares es un hallazgo inespecífico, sobretodo en protocolos agresivos Arroja resultados en un 40% Cuando se realiza un diagnóstico se realiza test genético dirigido confirmatorio Aunque las enfermedades identificadas se suelen tratar con DAI no está estuadiada la eficacia de esta medida, más en familiares identificados con test genéticos Se ha recomendado que los familiares de primer grado, en ausencia de cardiopatía estructural, sean remitidos a centros especializados. La realización de test genéticos de rutina no es practiable, salvo en diagnósticos específicos La detección de casos por estudio genético conllevaría , en el mejor de los casos, un seguimiento indefinido

40
Conclusiones La MS es una entidad frecuente
El cribado en familiares tiene un alto rendimiento (40%) Las causas principales: C. Isquémica e I. Cardíaca es fácilmente identificable y tratable Para las alteraciones primariamente eléctricas la solución suele ser colocación de DAI
 Las causas principales: C. Isquémica e I. Cardíaca es fácilmente identificable y tratable. Para las alteraciones primariamente eléctricas la solución suele ser colocación de DAI.")
Presentaciones similares








>")
















