Descargar la presentación
La descarga está en progreso. Por favor, espere

1
ANEMIA HEMOLITICA

2
Concepto Enfermedad hemolítica compensada

3
Clasificación Alteraciones Intrínsecas del hematíe
Defectos enzimáticos Hemoglobinopatías Intra cropusculares Hereditarias 2. Alteraciones de la membrana Esferocitosis hereditaria Hemoglobinuria paroxística nocturna Anemia con hematíes especulados Extra cropusculares 3. Factores extrínsecos Hiperesplenismo Hemolisis inmunitaria(por anticuerpos) Hemolisis microangopatica Infecciones toxinas Adquiridas
 Hemolisis microangopatica. Infecciones. toxinas. Adquiridas.")
4
oxidación cuando ocurre hemólisis intravascular la hemoglobina libre se disocia en dímeros a y b , los que se unen haptoglobina u oxidan a metahemoglobina. Luego estas moléculas se disocian y liberan el grupo Hemo para unirse a la albúmina y hemopexina, lo que permite la captación del Hemo por los hepatocitos para recuperar el fierro y para producir bilirrubina.

5
Hemólisis en laboratorio
Datos hematologicos Extravascular intravascular Frotis sanguíneo sistémico Policromatofilia Reticulocitos MO Proporcion mieloide/ eritoide Hiperplasia eritroide 1.5:1 Hiperplasia

6
Hemólisis en laboratorio
Plasma Extravascular Intravascular Bilirrubina indirecta Haptoglobina ausente DHL

7
Hemólisis en laboratorio
ORINA Extravascular Intravascular Bilirrubina Urobilinógeno + Hemosiderina Hemoglobina

8
Forma Eritrocitaria y Dx
Causa Sx Esferocitos Dianocitos Esquistocitos Hematies falciformes Perdida de la membrana Aumento del cociente superficie / volumen Ruptura traumática de la membrana Polimerizacion dela Hg S Esferocitosis hereditaria Autoinmune Hemoglobinopatías Procesos microangopaticos, protesis intravasculares Sx drepanocíticos

9
Forma Erirocitaria y Dx
Causa Sx Acantocitos Hematíes algutinados Cuerpos de Heinz Anormalidad de los lípidos de la membrana Anticuerpos IgM Hg precipitada Hepatopatías (anemias con hematíes espiculados) Enf por crioaglutininas Hemoglobinas inestables, agresión oxidante
 Enf por crioaglutininas. Hemoglobinas inestables, agresión oxidante.")
10
Anemias hemolíticas hereditarias
Defectos congénitos Membrana Enzimas hemoglobina

11
Anemias hemolíticas adquiridas
Hematíes formados con normalidad Destrucción prematura Lesiones adquiridas mientras circulan

13
Defectos Intracorpusculares
Defectos hereditarios de la membrana eritrocitaria La Esferocitosis hereditaria causa más frecuente de anemia hemolítica Defecto en proteínas del citoesqueleto Autosómica dominante 1:1000 a 1:4500

15
Esferocitosis hereditaria

16
Disminución de La relación Superficie volumen Defecto del primario citoesqueleto Aumento en la Inestabilidad de La membrana Disminución De la Deformidad celular Perdida de la membrana Atrapemiento esplénico Erirtoestasis Disminución de glucosa Disminución del ph Aum contacto con macrófagos Depleción de ATP Daño por acidez Hemólisis Fagocitosis Lisis osmótica Daño oxidativo

17
Hallazos Clínicos y de laboratorio
Manifestaciones Clínicas Anemia Esplenomegalia Ictericia Crisis aplásicas Crisis megaloblásticas Respuesta excelente a la esplenomegalia Ulcera crónica en tobillo

18
Hallazos Clínicos y de laboratorio
Reticulocitos mayor del 5 al 20% Esferocitos Aumento de la Concentración HCM (>36%) fragilidad osmótica aumentada Prueba de antiglobulina directa anormal
 fragilidad osmótica aumentada. Prueba de antiglobulina directa anormal.")
19
Clasificación clínica de Esferocitosis hereditaria
Portador silencioso leve Moderada Ligeramente grave grave Hg Reticulocitos Bilirrubina Extendido Periférico Fragilidad osmótica Fresca Incubada otros Nl 1-3 0-1 Ligeramente aumentada Padres de pacientes con EH no dominante 11-15 < 6 3-8 Pocos esferocitos Nl o Muy aumentada Hemólisis compensada 8-12 >8 >2 Esferocitosis evidente Hemólisis parcialmente compensada 6-8 >10 2-3 Esferocitosis Evidente Recesiva y dominante <6 >3 esferocitosis Dependiente de transfusiones

20
Tratamiento de esferocitosis hereditaria
Esplenectomía (5 a 9 años)
")
21
Defectos Intracorpusculares
Defectos hereditarios de la membrana eritrocitaria La Eliptocitosis hereditaria Eritrocitos elípticos u ovalados La forma de adquiere en la circulación Autosómica dominante 1:2000 a 1:4000

22
Eliptocitosis hereditaria
Defecto en espectrina y proteína 4.1 del citoesqueleto 9 tipos diferentes de defectos Disminución en vínculo de los dimeros de espectrina para formar tetradimeros debido a cadenas defectuosas Deficiencia o defecto en la banda 4.1 que ayuda al enlace de la espectrina con la actina Anormalidades de proteínas integrales

24
Morfología normal (87%) reticulocitos ligeramente aumentada (12%)
 reticulocitos ligeramente aumentada (12%)")
25
Dx Eliptocitosis hereditaria
examen morfológico de los eritrocitos circulantes en el paciente y sus familiares prueba de la fragmentación térmica eritrocitaria electroforesis de las proteínas del esqueleto de la membrana eritrocitaria PCR

26
Eliptocitosis

27
Defectos Intracorpusculares
Trastornos heredados de la permeabilidad a los cationes y del volumen eritocitario La Estomatocitosis hereditaria (hidrocitosis) Deficiencia de bomba Na:K Aumento de agua intracelular Autosómica dominante Rara
 Deficiencia de bomba Na:K. Aumento de agua intracelular. Autosómica dominante. Rara.")
28
Estomatocitosis hereditaria
Deficiencia de una proteína integral de la membrana llamada estomatina o banda 7.2b Entrada de Na supera la salida de K edema

29
Estomatocitosis

30
Otras causas Estomatosis adquirida Estomatosis en la enfermedad de
Rh null Estomatosis en enfermedad Rh mod

31
Trastornos heredados de la permeabilidad a los cationes y del volumen eritocitario
Xerocitosis hereditaria Rara Autosomica dominante Eritrocitos deshidratados Aumento de la HCM Se pierde K

32
Defectos Intracorpusculares
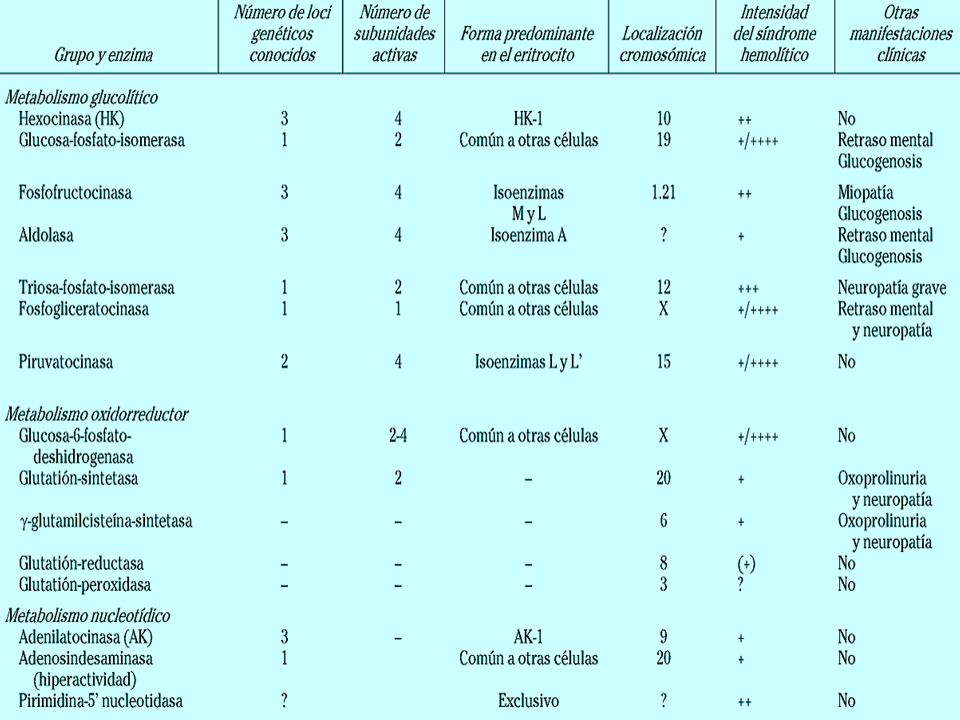
Enzimopatías eritrocitarias Deficiencia de glucosa 6 fosfato deshidrogenasa Vía oxidativa Recesivo ligado al cromosoma X Enzimopatía más frecuente Hombres homocigotos normales homocigotos deficientes Mujeres homocigotas normales homocigotas deficientes heterocigotos

33
Deficiencia de glucosa 6 fosfato deshidrogenasa
Clasificación de OMS Clase I: síntomas clínicos graves (anemia hemolítica crónica no esfeocitica) actividad de G6PD menor del 20% Clase II: expresión clínica leve, hemólisis intermitente y actividad menor al 10% Clase III: expresión clínica leve, hemolisis intermitente asociada con infecciones o fármacos, actividad del 10 al 60% Clase IV: No hay manifestaciones clínicas y actividad del 100%
 actividad de G6PD menor del 20% Clase II: expresión clínica leve, hemólisis intermitente y actividad menor al 10% Clase III: expresión clínica leve, hemolisis intermitente asociada con infecciones o fármacos, actividad del 10 al 60% Clase IV: No hay manifestaciones clínicas y actividad del 100%")
34
Deficiencia de glucosa 6 fosfato deshidrogenasa
G6FD Glucosa 6 fosfato + NADP fosfato gluconato

35
Deficiencia de glucosa 6 fosfato deshidrogenasa
Vulnerable al daño por oxidación Oxidación de tioles de la membrana Aparición de cuerpos de Heinz (precipitados intracelulares de Hg desnaturalizada) Anomalías del citoesqueleto que aumentan las perdidas de Na y K
 Anomalías del citoesqueleto que aumentan las perdidas de Na y K.")
36
Deficiencia de glucosa 6 fosfato deshidrogenasa
Anemia hemolítica como resultado a exposición a fármacos o infecciones Favismo Ictericia neonatal Anemia hemolítica crónica no esferocítica

38
Deficiencia de glucosa 6 fosfato deshidrogenasa
Salmonela Bacilos coliformes Estreptococo beta hemolítico Rickettsias Influenza Hepatitis virales

39
Deficiencia de glucosa 6 fosfato deshidrogenasa
actividad G-6-PD Tx Prevención Tratamiento ante infecciones Hemotransfusion si Hg disminuye entre 7 y 9 con evidencia de hemólisis continua

40
Enzimopatías eritrocitarias
Deficiencia de piruvato cinasa Vía glucolítica Defecto más común Autosómico recesivo disminución de la capacidad energética del eritrocito

41
Deficiencia de piruvato cinasa
intensa reticulocitosis alteraciones morfológicas de los eritrocitos son poco específicas vida media eritrocitaria determinar la actividad PK

42
Deficiencia de piruvato cinasa
Tx Tratamiento de sostén Transfusión en casos necesarios Esplenectomía Eleva 1 a 3 g de hg

44
Hemoglubinuria paroxística nocturna
Trastorno mieloproliferativo clonal de la medula ósea Anemia hemolítica intravascular Mayor susceptibilidad del eritrocito al complemento plaquetas y leucocitos Único trastorno hemolítico adquirido de membrana

45
Hemoglubinuria paroxística nocturna
Se carecen de CD55 (factor acelerador de la degranulación) CD59 (inhibidor de membrana de la lisis reactiva) Los complejos C3 convertasa no se eliminan
 CD59 (inhibidor de membrana de la lisis reactiva) Los complejos C3 convertasa no se eliminan.")
46
Forma complejos de ataque a la membrana con formación de poros y lisis celular
En la actualidad se ha detectado la disminución o desaparición de la expresión de más de veinte proteínas de membrana, que emplean el mismo sistema de anclaje

47
Clínica anemia hemolítica crónica con ocasionales episodios de agudización nocturna, coincidiendo con infecciones o con la administración de medicamentos o transfusiones trombosis de territorios venosos defecto hemopoyético que puede cursar con pancitopenia posible asociación de un defecto inmune con infecciones de repetición

48
DX anemia, leucopenia y trombocitopenia de intensidad variable
reticulocitos, elevación de LDH y de bilirrubina y descenso de la cifra de haptoglobina Hemosiderinuria hiperplasia de la serie roja o aplasia global hemólisis eritrocitaria provocada en un medio acidificado identificación de los defectos proteicos CD55 y CD59 (serie roja), CD66/67 para los granulocitos y CD14, CD55 y CD59 para los monocitos
, CD66/67 para los granulocitos y CD14, CD55 y CD59 para los monocitos.")
49
Tx Transfusiones Anticoagulantes Tratamiento con hierro Esteroide
Transplante de MO

50
Defectos extracropusculares no autoinmunes
Anemia Hemolítica microagiopática Grupo de enfermedades caracterizado por fragmentación de los eritrocitos en la circulación que produce hemólisis intravascular

51
Anemia Hemolítica microagiopática
Púrpura trombótica trombocitopénica Sx urémico-hemolítico

52
Púrpura trombótica trombocitopénica
Sx. clínico caracterizado por: Anemia hemolítica con fragmentación de eritrocitos Plaquetopenia Disfunción neurológica Fiebre Insuficiencia renal progresiva Oclusiones trombóticas en la microcirculación

53
Sx urémico-hemolítico
Sx caracterizado por: Anemia hemolítica Plaquetopenia Insuficiencia renal grave Asociada a infección entérica Verocitotoxina producida por E coli

54
Anemia Hemolítica microagiopática
Hipertensión maligna Coagulación intravascular diseminada Carcinoma diseminado

55
Anemia hemolítica macrovascular
Anemia hemolítica cardiaca traumática Hemoglobinuria de la marcha

56
Defectos extracropusculares de causa inmune
Alteración de los eritrocitos resultado de un episodio inmune que se produce en la superficie del eritrocito

57
Defectos extracropusculares de causa inmune

58
Reacciones hemolíticas postransfusionales
se producen cuando se transfunden hematíes que contienen antígenos para los cuales el receptor tiene anticuerpos. Éstos pueden ser naturales (sistema ABO) o inmunes (sistema Rh y Kell, entre otros).
 o inmunes (sistema Rh y Kell, entre otros).")
59
Reacciones hemolíticas postransfusionales
Puede manifestarse por: escalofríos Hipertermia dolor lumbar Hipotensión Shock insuficiencia renal.

60
Reacciones hemolíticas postransfusionales
Dx LDH sérica descenso de la haptoglobina hemoglobinemia hemoglobinuria.

61
Enfermedad hemolítica del recién nacido
existe incompatibilidad entre los antígenos eritrocitarios de la madre y los del feto. ejemplo clásico es la isoinmunización por el antígeno D del sistema Rh cualquier antígeno de grupo sanguíneo ausente en la madre y presente en el feto puede inducir la formación de aloanticuerpos que causen la hemólisis neonatal.

62
Enfermedad hemolítica del recién nacido
La mujer puede entrar en contacto por primera vez con el antígeno por una transfusión o por un embarazo. Cuando se produce el segundo contacto con el antígeno, habitualmente en el segundo embarazo, los anticuerpos de clase IgG (especialmente IgG3 o IgG1) desarrollados en la madre atraviesan la placenta y se fijan a los hematíes del feto portadores del antígeno correspondiente, produciendo su hemólisis
 desarrollados en la madre atraviesan la placenta y se fijan a los hematíes del feto portadores del antígeno correspondiente, produciendo su hemólisis.")
63
Enfermedad hemolítica del recién nacido
En el momento de nacer, la prueba de Coombs directa sobre los hematíes del recién nacido prueba de Coombs indirecta en el suero de la madre permite establecer el diagnóstico diferencial con otras ictericias neonatales.

64
Anemia hemolítica Autoinmune
Destrucción prematura de los eritrocitos debido a la presencia de autoanticuerpos que se unen a los antígenos presentes en la superficie de los eritrocitos Perdida de la tolerancia inmune

Presentaciones similares












>")











. Papel biológico: No se conoce. Importantes en : Genética Medicina legal.>")