Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Alteraciones Mec. Intrínseco Congénitas - Adquiridas

2
Congénitas Adquiridas COAGULOPATIAS

3
HEMOFILIA ENFERMEDAD DE VON WILLEBRAND

4
PREVALENCIA - Hemofilia A1/5000 - Hemofilia B1/25000 - VWD 1% población

5
IIa A1A2 B A3 C1 C2 200kD 80kD 200kD 80kD 90kD HRG 80kD 50kD 43kD 73kD 45kD 67kD FACTOR VIII NH 2 SecreciónPreactivaciónActivaciónInactivación IIaIIa APC IIa Xa Xa 1648 740 372 336 1689 1721 Lollar P & Parker CG, 1989

6
FVIII-VWF S-SS-S VWF FVIII FVIIIa IIa, FXa A1 A2 C2 C1 A3 N N C C 1689 740 372 A1 A2 A3 C1 C2 B Vlot et al., 1998

7
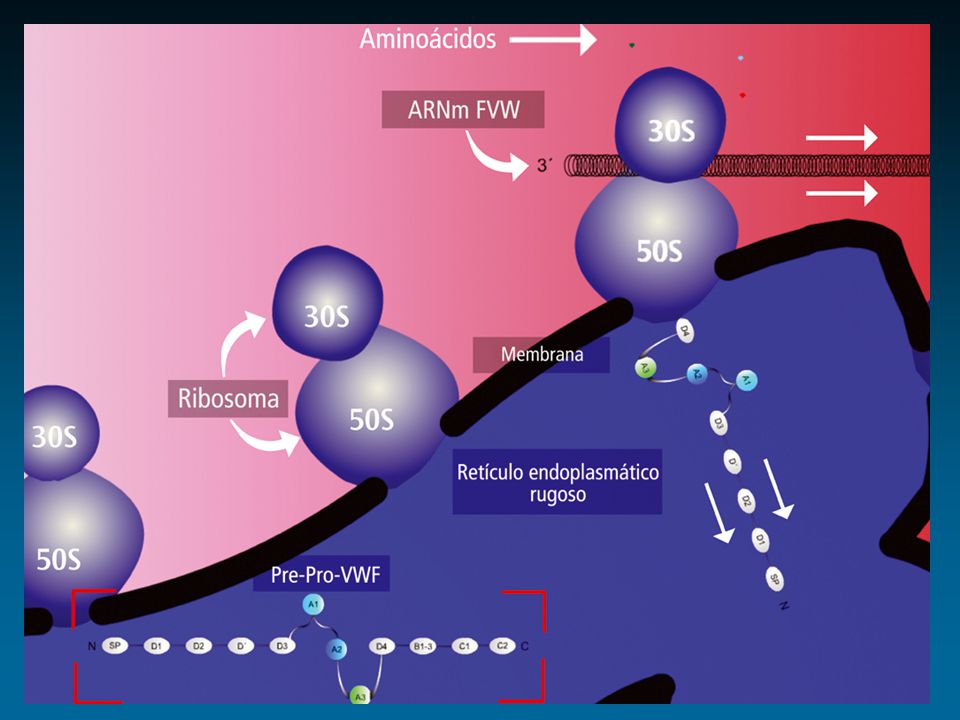
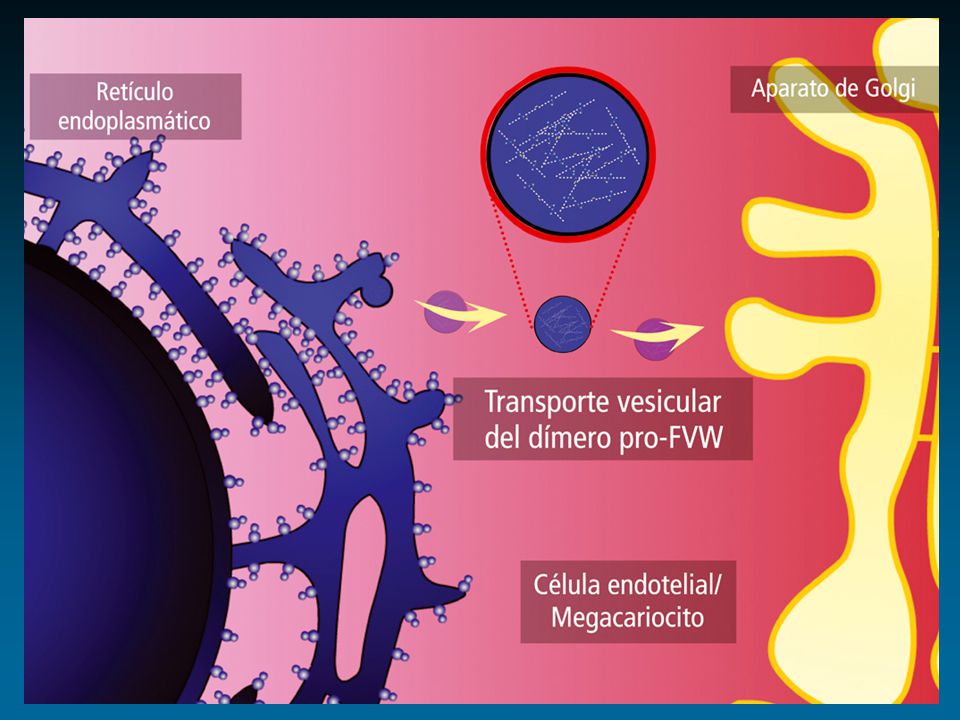
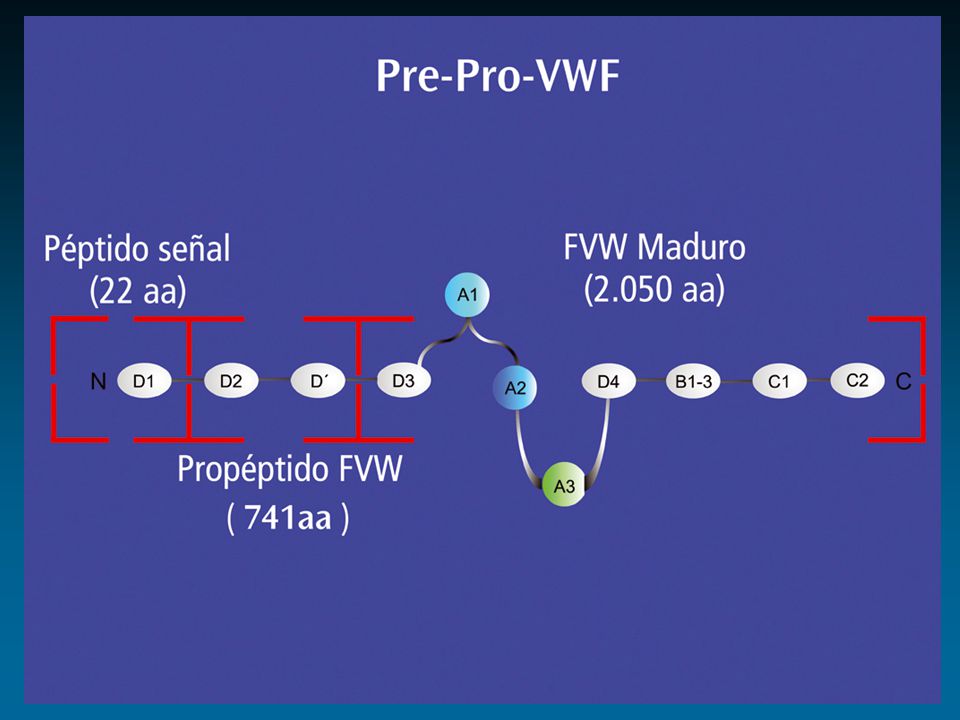
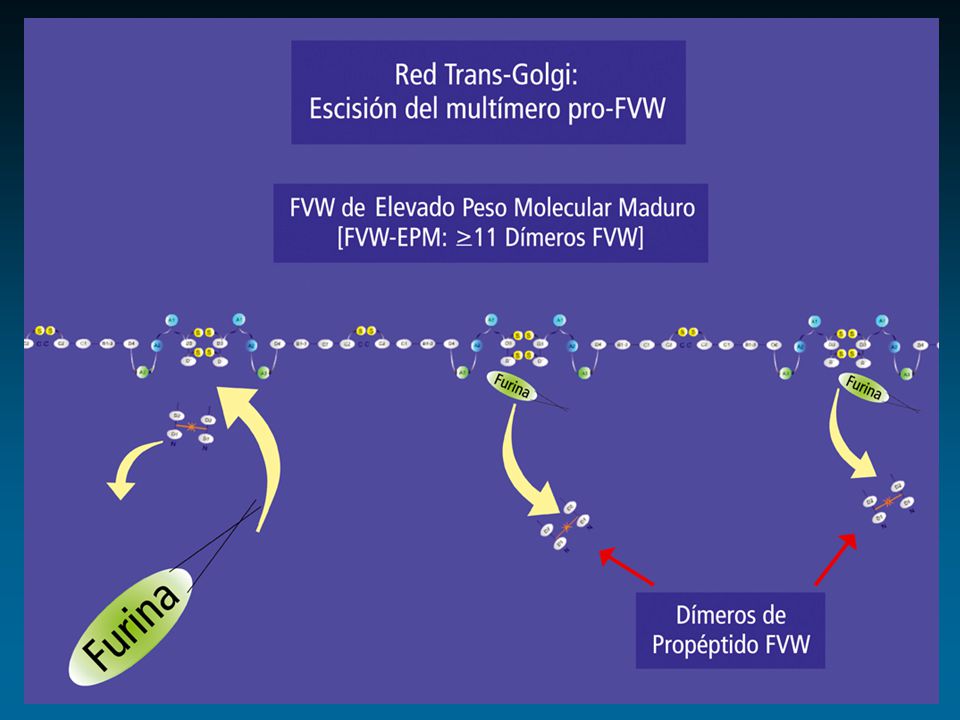
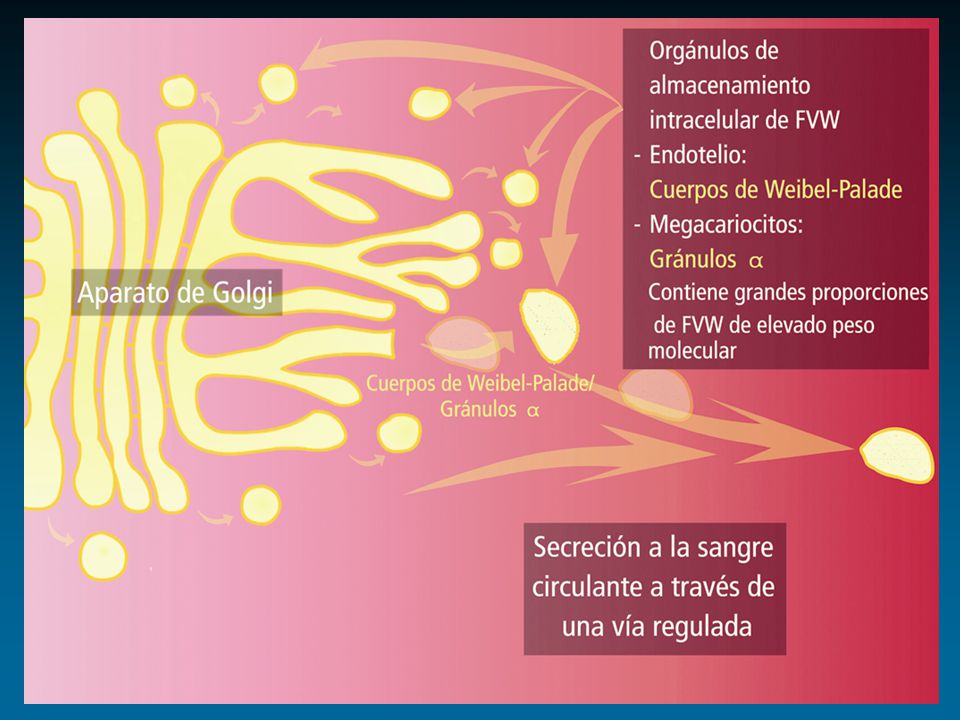
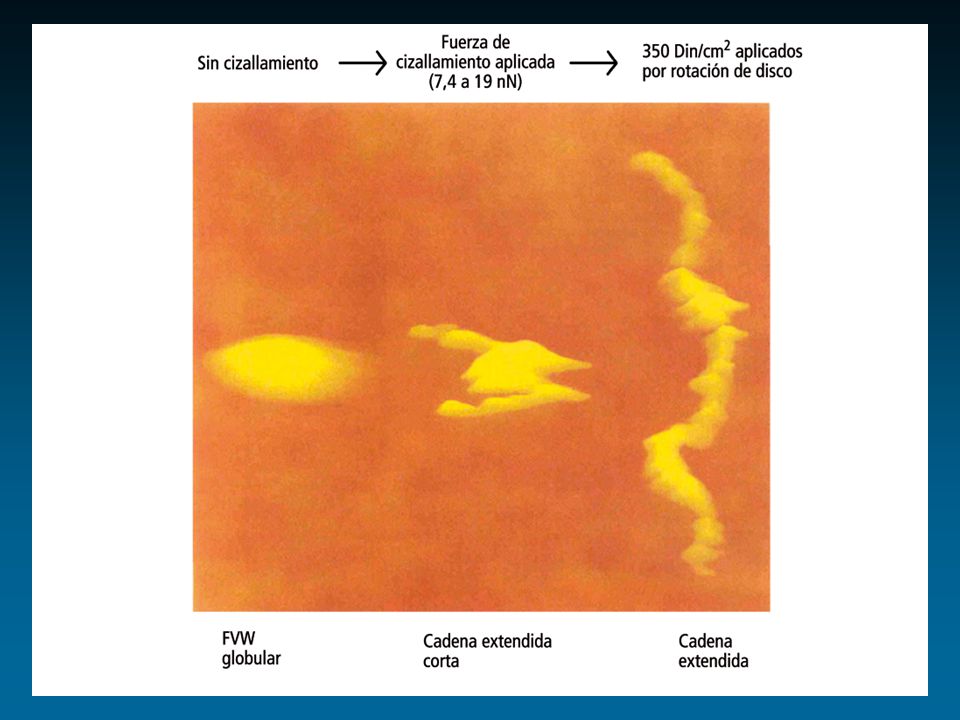
Factor de von Willebrand: Cr 12

14
ADAMTS 13

15
Sadler JE, 2005 vWF Ensamble y Catabolismo Proteolisis aumentada Depuración aumentada Plasma Inicial k proteolisis k depuración ADAMTS-13 k secreción

17
FVIII/VWF - Propiedades FVIII VWF ClínicaHemofilia vWD ProducciónS. Hepáticos C. Endoteliales Megacariocitos Megacariocitos GenCromosoma X Cromosoma 12 MoléculaHeterodímero Multímero FunciónCoagulación Cofactor Adhesión Adhesión Endotelio/Sub Endotelio/Sub Transporta FVIII Transporta FVIII

18
FVIII/VWF - Nomenclatura Factor VIII Antígeno FVIII:Ag Función FVIII Factor von Willebrand Proteína Madura VWF Antígeno VWF:Ag Cofactor Ristocetina VWF:RCo Unión al Colágeno VWF:CB Unión al Factor VIII VWF:FVIIIB ISTH

19
Recesiva Ligada al sexo (gen FVIII Cr X) PORTADORA HEMOFILIA A HEMOFILICO
 PORTADORA HEMOFILIA A HEMOFILICO")
20
HEMOFILIA Modo de Herencia

21
HEMOFILIA

22
q28 Cromosoma X Factor VIIIG6PD F8A F8B Exon 22 Exon 23 5’3’ GEN FACTOR VIII 1 2 3 4 5 6 7 8 9 10 1112 13 14 1516171819 20 21 22 23 24 25 26

24
HEMOFILIA Diagnóstico “sencillo” - APTT prolongado - FVIII disminuido

25
HEMOFILIA - Clasificación Severa< 1% VIII Moderada1-5% VIII Leve>5% VIII

26
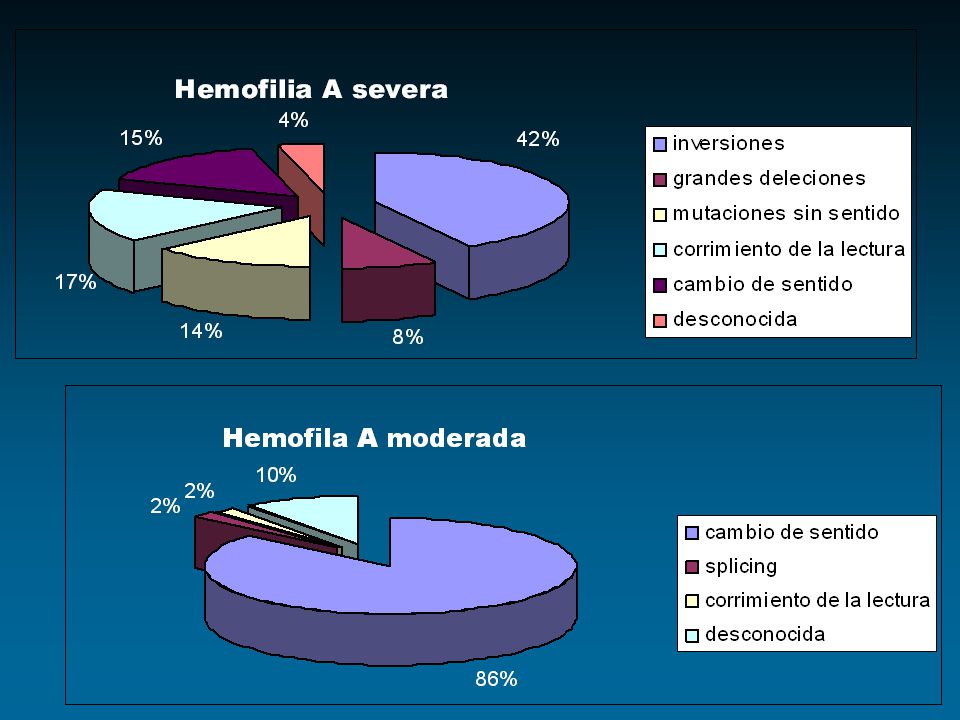
Alteración heterogénea Alteración heterogénea Afecta diferentes regiones Afecta diferentes regiones del gen No aplicable a todos los casos No aplicable a todos los casos Análisis de ADN Complejo

27
Inversión del intrón 22 ↓ 50% hemofílicos severos IDENTIFIACION DIRECTA Certeza

28
Inversiones Inversiones Deleciones Deleciones Inserciones Inserciones Mutaciones “sin sentido” Mutaciones “sin sentido” Mutaciones “sentido falso” Mutaciones “sentido falso” IDENTIFIACION DIRECTA

29
Recesiva Ligada al sexo (gen FIX Cr X) PORTADORA HEMOFILIA B HEMOFILICO
 PORTADORA HEMOFILIA B HEMOFILICO")
30
HEMOFILIA - Clasificación Severa< 1% IX Moderada1-5% IX Leve>5% IX

31
HEMOFILIA B Diagnóstico “sencillo” - APTT prolongado - FIX disminuido

32
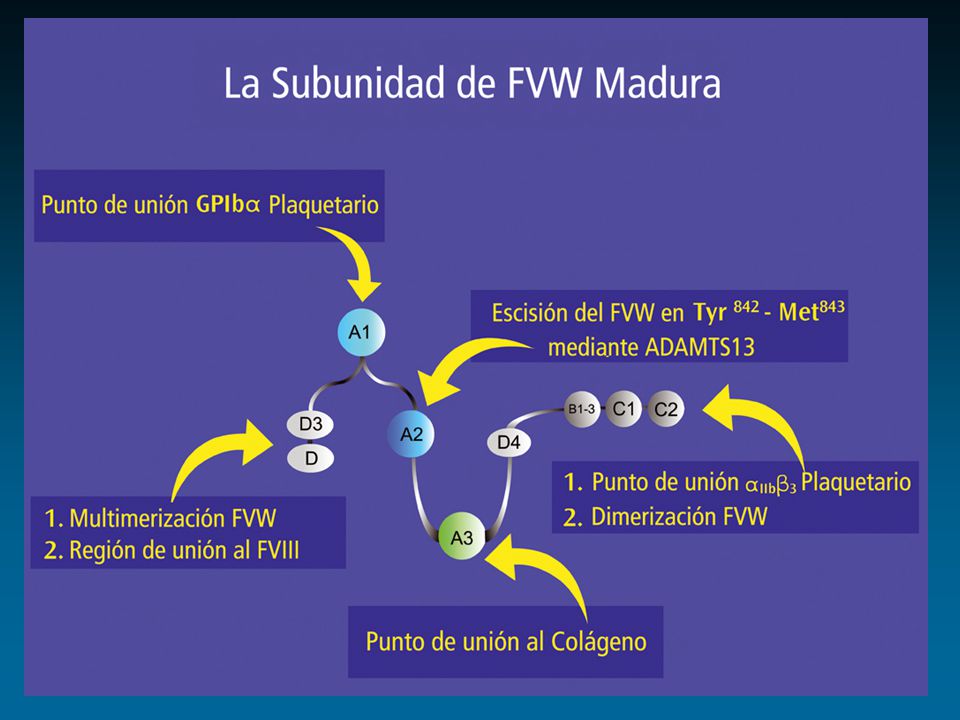
protómero dominios funcionales ligandos Colágeno GPIIb-IIIa FVIII GPIb Heparina Heparina Colágeno Sulfátidos D 1 D 2 D’ D 3 A 1 A 2 A 3 D 4 BC 1 C 2 D’ D 3 A 1 A 2 A 3 D 4 BC 1 C 2 1 2050 S I S I S RGD RGD Enfermedad de von Willebrand Propéptido Subunidad Madura

33
Sadler JE, 2005 vWF Ensamble y Catabolismo Proteolisis aumentada (vWD2A) Depuración aumentada (vWD1 Vicenza) Plasma Inicial k proteolisis k depuración ADAMTS-13 k secreción
 Depuración aumentada (vWD1 Vicenza) Plasma Inicial k proteolisis k depuración ADAMTS-13 k secreción")
34
VWF Expresión Variable Penetrancia Incompleta Chance Modificadores Ambientales edad, género, trauma Enfermedad primaria mutación ? Genes modificadores Ginsburg D, 2001 Difícil Diagnóstico Niveles Variables VWF

35
VWD Clasificación Revisada Sadler E, 1994 Tipo 1Deficiencia cuantitativa parcial Tipo 2Alteración cualitativa Tipo 3“Ausencia”de VWF

36
VWD – TIPOS Frecuencia (%) TIPO 1 TIPO 2 TIPO 3 Tuddenham (134) 75196 Lenk (111) 761212 Nilson (106f) 701020 Hoyer (116) 71236 Awidi (65) 5929,511,5 Berliner (60) 62929 Castaman G, 2003
 TIPO 1 TIPO 2 TIPO 3 Tuddenham (134) Lenk (111) Nilson (106f) Hoyer (116) Awidi (65) 5929,511,5 Berliner (60) Castaman G, 2003")
37
VWD Serie 1885 Pacientes (Tipo I 91%) FVIII 61,6% FVIII 61,6% VWF:Ag62,2% VWF:Ag62,2% TS 58,4% TS 58,4% Woods AI, 2001
 FVIII 61,6% FVIII 61,6% VWF:Ag62,2% VWF:Ag62,2% TS 58,4% TS 58,4% Woods AI, 2001")
38
Individual Individual Familiar Familiar VWF-Variabilidad

39
VWF-Variabilidad Factores 40% no genéticos 40% no genéticos 60% genéticos 60% genéticos

40
VWF-Variabilidad Factores no-genéticos Edad Edad Estrés Estrés Ejercicio Ejercicio H. Tiroideas H. Tiroideas Estrógenos Estrógenos F. Aguda F. Aguda

41
VWF-Variabilidad Factores genéticos Grupo ABO 20-30% variabilidad Grupo ABO 20-30% variabilidad Grupo “O” VWF Hipótesis Glicosilación Alteración procesamiento procesamiento estabilidad estabilidad secreción secreción

42
VWD Distribución según grupo ABO “AB” 0,6% difícil diagnóstico “AB” 0,6% difícil diagnóstico “O” 70% más sintomático “O” 70% más sintomático > frecuencia de consulta Woods AI, 2001

43
Cuantitativa Cualitativa VWF:Ag SI NO VWF:RCo SI SI VWF:CB SI SI Multímeros SEMI SI VWF Pruebas Específicas

44
VWFpp-ELISA VWFpp Síntesis VWF (independiente ABO) (independiente ABO) Tipo 1VWFpp Tipo 1VWFpp Tipo 3VWFpp ausente Tipo 3VWFpp ausente
 (independiente ABO) Tipo 1VWFpp Tipo 1VWFpp Tipo 3VWFpp ausente Tipo 3VWFpp ausente")
45
VWF:RCo Plaquetasnormales Plasma del paciente Ristocetina Plaquetas del paciente Plasma Ristocetina RIPA Agreg. c/Ristocetina Agregometría Agregometría PRP

46
Agregación c/Ristocetina Kasper C, 2005 Normal vWD-Tipo 1 vWD-Tipo 2A vWD-Tipo 3

47
2A función plaquetaria Multímeros APM e intermedio vWD-Tipo 2 Clasificación Revisada Sadler E, 1994

48
Normal 2A Línea 1 Línea 2 Línea 3 Lane 2> Lane 1> Lane 3> Línea 2= 95% Línea 3= 3% VWF-Análisis Multimérico grandes intermedios-pequeños (banda 1)(banda 2)
(banda 2)")
49
2A función plaquetaria Multímeros APM e intermedio vWD-Tipo 2 Clasificación Revisada Sadler E, 1994 2B afinidad VWF-GPIb-IX

50
Agregación c/Ristocetina Kasper C, 2005

51
2B Normal 2A Línea 1 Línea 2 Línea 3 Lane 3> VWF-Análisis Multimérico grandes intermedios-pequeños (banda 1)(banda 2)
(banda 2)")
52
2A función plaquetaria Multímeros APM e intermedio vWD-Tipo 2 Clasificación Revisada Sadler E, 1994 2B afinidad VWF-GPIb-IX 2M función plaquetaria c/Multímeros APM 2N afinidad VWF-FVIII* * símil Hemofilia A, autosómica

53
protómero dominios funcionales ligandos Colágeno GPIIb-IIIa FVIII GPIb Heparina Heparina Colágeno Sulfátidos D 1 D 2 D’ D 3 A 1 A 2 A 3 D 4 BC 1 C 2 D’ D 3 A 1 A 2 A 3 D 4 BC 1 C 2 1 2050 S I S I S RGD RGD Factor von Willebrand Propéptido Subunidad Madura X

54
2N-Variante Normandy Biología Molecular -Secuenciación -Enz. restricción

55
VWF- Arg19Trp (C2594T) Exón 18 Enzima BspMI 1-244 pb 2-148 pb 3-96 pb Normal 148 pb 96 pb Mutado 244 pb 123 MutaciónNormal
 Exón 18 Enzima BspMI pb pb 3-96 pb Normal 148 pb 96 pb Mutado 244 pb 123 MutaciónNormal")
56
T. Sangría T. Sangría APTT (fVIII) APTT (fVIII) Adhesividad Adhesividad Tiempo de obstrucción (PFA) Tiempo de obstrucción (PFA) Screening VWD Estrategia Diagnóstica * *no permiten excluir VWD
 APTT (fVIII) Adhesividad Adhesividad Tiempo de obstrucción (PFA) Tiempo de obstrucción (PFA) Screening VWD Estrategia Diagnóstica * *no permiten excluir VWD.")
57
-VWF plasma/plaquetas VWD Estrategia Diagnóstica Diagnóstico VWF:Ag VWF:Ag VWF:RCo VWF:RCo VWF:CB VWF:CB -FVIII en plasma

58
Multímeros Multímeros RIPA RIPA Unión GP plaquetarias Unión GP plaquetarias VWF:FVIIIB VWF:FVIIIB Biología Molecular Biología Molecular VWD Estrategia Diagnóstica Clasificación

59
vWD- Importancia Dignóstica VWF:RCo Prueba más sensible VWF:Ag Correlaciona VWF:RCo Ratio=1 Tipo1 Ratio<1 Tipo2 FVIII Util para el manejo clínico T. Sangría Correlaciona VWF:RCo (T1) Detecta defecto plaquetario RIPA Detecta Tipo 2B Castaman G, 2003
 Detecta defecto plaquetario RIPA Detecta Tipo 2B Castaman G,")
60
vWD- Importancia Dignóstica A.Multimérico Discrimina Tipo 2M, 2A, 2B VWF plaquetario Predice respuesta DDAVP Tipo 1 VWF:FVIIIB Detecta Tipo 2N VWF:CB Correlaciona VWF:RCo (T1) Sensibilidad colágeno CT PFA-100 Especificidad (?) Castaman G, 2003
 Sensibilidad colágeno CT PFA-100 Especificidad ( ) Castaman G, 2003")
61
HEMOFILIA VON WILLEBRAND Diagnóstico Diferencial

62
Cofactor de ristocetina * Cofactor de ristocetina * Antígeno de factor vW o N Antígeno de factor vW o N Factor VIII o N* Factor VIII o N* Tiempo de Sangría o N Tiempo de Sangría o N *excepto la variante 2N Enfermedad de von Willebrand

63
Cofactor de ristocetinaN Cofactor de ristocetinaN Antígeno de factor vW N Antígeno de factor vW N Factor VIII Factor VIII Tiempo de Sangría N Tiempo de Sangría N Hemofilia

64
Hemofilia VWD2N Hemofilia VWD2N VWF:RCo N N Factor VIII VWF:FVIIIB N Hemofilia-VWD 2N

65
Fragmentos de degradación (intraplaquetario normal) Fragmentos de degradación (intraplaquetario normal) VWFpp depuración VWFpp depuración aVWF (inhibición VWF:RCo, VWF:Ag, FVIII) aVWF (inhibición VWF:RCo, VWF:Ag, FVIII) VWD - Adquirido Laboratorio Mecanismos
 Fragmentos de degradación (intraplaquetario normal) VWFpp depuración VWFpp depuración aVWF (inhibición VWF:RCo, VWF:Ag, FVIII) aVWF (inhibición VWF:RCo, VWF:Ag, FVIII) VWD - Adquirido Laboratorio Mecanismos")
66
OTROS DESORDENES HEREDITARIOS

67
DESORDENES HEREDITARIOS-Recesivos Frecuencia Depende del tipo población Grado de cosanguinidad

68
DEFICIT DE FACTOR XII -Generalmente, no asociado a desórdenes hemorrágicos -IAM, TEP -Menorragias, hemorragias subaracnoideas

69
DEFICIT PRECALICREINA - Sin complicaciones hemorrágicas - Riesgo de IAM, TEP, stroke

70
DEFICIT PRECALICREINA Diagnóstico - TTPA -Corrige con normal -Corrige con incubación prolongada (10 min a 37º C) - PK (actividad coagulante)
 - PK (actividad coagulante)")
71
DEFICIT PRECALICREINA Diagnóstico - TTPA -Corrige con normal -Corrige con incubación prolongada (10 min a 37º C) - PK (actividad coagulante)
 - PK (actividad coagulante)")
72
DEFICIT DE QUININOGENOS DE ALTO PESO MOLECULAR Sin asociación con desórdenes hemorrágicos Riesgo de TVP y TEP

73
FACTOR XI Vida media en plasma: 50-80 hs. Nivel hemostático: 15-20% Déficit Prevalencia: 1 en 100.000 (población general) 1 en 190 (judíos Ashkenazi)
 1 en 190 (judíos Ashkenazi).")
74
DEFICIT DE FACTOR XI Clínica -Sangrado espontáneo raro -Hematomas fáciles, sangrado post extracción dentaria, cirugía, parto -Sangrado en sitios ↑ Fibrinolisis (cavidad oral, nariz y útero)
")
75
DEFICIT DE FACTOR XI Clínica ¿Por qué sangra sólo el 50% de los pacientes y sin relación con los niveles plasmáticos de FXI?

76
Congénitas Adquiridas COAGULOPATIAS

77
Adquiridas DéficitInhibición

78
Neutralizantes EF Ac No neutralizantes Ac EF ESPECIFICOS INHIBIDORES ADQUIRIDOS

79
ESPECIFICOSESPECIFICOS Neutralizantes INHIBEN ACTIVIDAD Afectan las pruebas de la coagulación EF Ac

80
ANTICUERPOS aFVIII ALOANTICUERPOS - Respuesta inmune a proteínas exógenas (terapéutica) AUTOANTICUERPOS - Autoinmunidad (neo-epitopes) ALOANTICUERPOS - Respuesta inmune a proteínas exógenas (terapéutica) AUTOANTICUERPOS - Autoinmunidad (neo-epitopes)
 AUTOANTICUERPOS - Autoinmunidad (neo-epitopes) ALOANTICUERPOS - Respuesta inmune a proteínas exógenas (terapéutica) AUTOANTICUERPOS - Autoinmunidad (neo-epitopes)")
81
XIIXIIa XIXIa IXIXa FL Ca 2+ IXIXa FL Ca 2+ FCFC XXaVaFL XXaVaFL II IIIIa IIa Fibrinógeno Fibrina FibrinaFibrinógeno HEMOFILIA A Tratamiento Tratamiento FVIIIFVIII

82
XIIXIIa XIXIa IXIXa FVIIIa FVIIIaFL Ca 2+ IXIXa FVIIIa FVIIIaFL Ca 2+ FCFC XXaVaFL XXaVaFL II IIIIa IIa Fibrinógeno Fibrina FibrinaFibrinógeno a-FVIIIa-FVIII aparecen durante el aparecen durante el 1 er año de tratamiento aparecen durante el aparecen durante el 1 er año de tratamiento INHIBIDORES aFVIII

83
INHIBIDORES aFVIII (aFVIII neutralizantes) CLINICA Hemorrágica aFVIII Complican la terapéutica sustitutiva post-cirugía/trauma Hemofílicos
 CLINICA Hemorrágica aFVIII Complican la terapéutica sustitutiva post-cirugía/trauma Hemofílicos")
84
ANTICUERPOS aFVIII AUTOANTICUERPOS Hemofilia Adquirida AUTOANTICUERPOS Hemofilia Adquirida

85
ANTICUERPOS aFVIII Hemofilia Adquirida Embarazo-Parto Enf. Autoinmune Neoplasia Idiopáticos (Edad ) Hemofilia Adquirida Embarazo-Parto Enf. Autoinmune Neoplasia Idiopáticos (Edad ) Cohen AJ & Kessler CM, 1996
 Hemofilia Adquirida Embarazo-Parto Enf. Autoinmune Neoplasia Idiopáticos (Edad ) Cohen AJ & Kessler CM,")
86
Inhibición tiempo y temperatura-dependiente aFVIII Neutralizantes Característica

87
Detección

88
APTT prolongado APTT prolongado No corrige con plasma normal No corrige con plasma normal Potencia con incubación Potencia con incubación No aumenta la actividad por dilución No aumenta la actividad por dilución aFVIII Neutralizantes Detección

89
Incubación (1 h a 37°C) P : 75 seg N : 32 seg P+N inm : 50 seg P+N inc : 70 segP+N inc : 50 seg POTENCIANO POTENCIA Incubación (1 h a 37°C) P : 75 seg N : 32 seg P+N inm : 50 seg P+N inc : 70 segP+N inc : 50 seg POTENCIANO POTENCIA INHIBICION TIEMPO DEPENDIENTE INHIBICION TIEMPO DEPENDIENTE
 P : 75 seg N : 32 seg P+N inm : 50 seg P+N inc : 70 segP+N inc : 50 seg POTENCIANO POTENCIA Incubación (1 h a 37°C) P : 75 seg N : 32 seg P+N inm : 50 seg P+N inc : 70 segP+N inc : 50 seg POTENCIANO POTENCIA INHIBICION TIEMPO DEPENDIENTE INHIBICION TIEMPO DEPENDIENTE")
90
INHIBIDORES DE INTERFERENCIA Inhibidor Lúpico Paraproteínas PDF/pdf Heparina-Heparinoides Inhibidor Lúpico Paraproteínas PDF/pdf Heparina-Heparinoides

91
COAGULPATIAS Orientación Diagnóstica

92
XII XIIa XI XIa IX IXa VIIIaFL Ca 2+ FC XXaVaFL IIa IIa Fibrinógeno M. Fibrina M. Fibrina Fibrina FibrinaFibrinógeno M. Fibrina M. Fibrina Fibrina Fibrina FTVIIa Ca 2+ XIIIa Activación II V. INTRINSECA TTPA V. EXTRINSECA TP V. FINAL TT TT PRUEBAS

93
INTERPRETACION Pruebas DEFECTO (probable) TP APTT, TT, TS Normales V. Extrínseca (FVII) Déficit de factores Vit K Dep. Hepatopatía TP, APTT TT, TS Normales V. Común, Déficit de factores Vit K Dep., Hepatopatía TP, APTT, TT, TS Hepatopatía, CID ↑ TP, APTT, TT TS Normal V. Final Fibrinógeno APTT TP, TT, TS Normal V. Intrínseca (FVIII, FIX, FXI, FXII) Hemofilia, VWD ↑ APTT, TS TP, TT Normales VWD
 Déficit de factores Vit K Dep. Hepatopatía TP, APTT TT, TS Normales V. Común, Déficit de factores Vit K Dep., Hepatopatía TP, APTT, TT, TS Hepatopatía, CID ↑ TP, APTT, TT TS Normal V. Final Fibrinógeno APTT TP, TT, TS Normal V. Intrínseca (FVIII, FIX, FXI, FXII) Hemofilia, VWD ↑ APTT, TS TP, TT Normales VWD.")
94
INTERPRETACION Pruebas DEFECTO (probable) TP APTT, TT, TS Normales V. Extrínseca (FVII) Déficit de factores Vit K Dep. Hepatopatía TP, APTT TT, TS Normales V. Común, Déficit de factores Vit K Dep., Hepatopatía TP, APTT, TT, TS Hepatopatía, CID ↑ TP, APTT, TT TS Normal V. Final Fibrinógeno APTT TP, TT, TS Normal V. Intrínseca (FVIII, FIX, FXI, FXII) Hemofilia, VWD ↑ APTT, TS TP, TT Normales VWD
 Déficit de factores Vit K Dep. Hepatopatía TP, APTT TT, TS Normales V. Común, Déficit de factores Vit K Dep., Hepatopatía TP, APTT, TT, TS Hepatopatía, CID ↑ TP, APTT, TT TS Normal V. Final Fibrinógeno APTT TP, TT, TS Normal V. Intrínseca (FVIII, FIX, FXI, FXII) Hemofilia, VWD ↑ APTT, TS TP, TT Normales VWD.")
95
INTERPRETACION Pruebas DEFECTO (probable) TP APTT, TT, TS Normales V. Extrínseca (FVII) Déficit de factores Vit K Dep. Hepatopatía TP, APTT TT, TS Normales V. Común, Déficit de factores Vit K Dep., Hepatopatía TP, APTT, TT, TS Hepatopatía, CID ↑ TP, APTT, TT TS Normal V. Final Fibrinógeno APTT TP, TT, TS Normal V. Intrínseca (FVIII, FIX, FXI, FXII) Hemofilia, VWD ↑ APTT, TS TP, TT Normales VWD
 Déficit de factores Vit K Dep. Hepatopatía TP, APTT TT, TS Normales V. Común, Déficit de factores Vit K Dep., Hepatopatía TP, APTT, TT, TS Hepatopatía, CID ↑ TP, APTT, TT TS Normal V. Final Fibrinógeno APTT TP, TT, TS Normal V. Intrínseca (FVIII, FIX, FXI, FXII) Hemofilia, VWD ↑ APTT, TS TP, TT Normales VWD.")
96
INTERPRETACION Pruebas DEFECTO (probable) TP APTT, TT, TS Normales V. Extrínseca (FVII) Déficit de factores Vit K Dep. Hepatopatía TP, APTT TT, TS Normales V. Común, Déficit de factores Vit K Dep., Hepatopatía TP, APTT, TT, TS Hepatopatía, CID ↑ TP, APTT, TT TS Normal V. Final Fibrinógeno APTT TP, TT, TS Normal V. Intrínseca (FVIII, FIX, FXI, FXII) Hemofilia, VWD ↑ APTT, TS TP, TT Normales VWD
 Déficit de factores Vit K Dep. Hepatopatía TP, APTT TT, TS Normales V. Común, Déficit de factores Vit K Dep., Hepatopatía TP, APTT, TT, TS Hepatopatía, CID ↑ TP, APTT, TT TS Normal V. Final Fibrinógeno APTT TP, TT, TS Normal V. Intrínseca (FVIII, FIX, FXI, FXII) Hemofilia, VWD ↑ APTT, TS TP, TT Normales VWD.")
97
INTERPRETACION Pruebas DEFECTO (probable) TP APTT, TT, TS Normales V. Extrínseca (FVII) Déficit de factores Vit K Dep. Hepatopatía TP, APTT TT, TS Normales V. Común, Déficit de factores Vit K Dep., Hepatopatía TP, APTT, TT, TS Hepatopatía, CID ↑ TP, APTT, TT TS Normal V. Final Fibrinógeno APTT TP, TT, TS Normal V. Intrínseca (FVIII, FIX, FXI, FXII) Hemofilia, VWD ↑ APTT, TS TP, TT Normales VWD
 Déficit de factores Vit K Dep. Hepatopatía TP, APTT TT, TS Normales V. Común, Déficit de factores Vit K Dep., Hepatopatía TP, APTT, TT, TS Hepatopatía, CID ↑ TP, APTT, TT TS Normal V. Final Fibrinógeno APTT TP, TT, TS Normal V. Intrínseca (FVIII, FIX, FXI, FXII) Hemofilia, VWD ↑ APTT, TS TP, TT Normales VWD.")
98
INTERPRETACION Pruebas DEFECTO (probable) TP APTT, TT, TS Normales V. Extrínseca (FVII) Déficit de factores Vit K Dep. Hepatopatía TP, APTT TT, TS Normales V. Común, Déficit de factores Vit K Dep., Hepatopatía TP, APTT, TT, TS Hepatopatía, CID ↑ TP, APTT, TT TS Normal V. Final Fibrinógeno APTT TP, TT, TS Normal V. Intrínseca (FVIII, FIX, FXI, FXII) Hemofilia, VWD ↑ APTT, TS TP, TT Normales VWD
 Déficit de factores Vit K Dep. Hepatopatía TP, APTT TT, TS Normales V. Común, Déficit de factores Vit K Dep., Hepatopatía TP, APTT, TT, TS Hepatopatía, CID ↑ TP, APTT, TT TS Normal V. Final Fibrinógeno APTT TP, TT, TS Normal V. Intrínseca (FVIII, FIX, FXI, FXII) Hemofilia, VWD ↑ APTT, TS TP, TT Normales VWD.")
99
INTERPRETACION Pruebas DEFECTO (probable) TP APTT, TT, TS Normales V. Extrínseca (FVII) Déficit de factores Vit K Dep. Hepatopatía TP, APTT TT, TS Normales V. Común, Déficit de factores Vit K Dep., Hepatopatía TP, APTT, TT, TS Hepatopatía, CID ↑ TP, APTT, TT TS Normal V. Final Fibrinógeno APTT TP, TT, TS Normal V. Intrínseca (FVIII, FIX, FXI, FXII) Hemofilia, VWD ↑ APTT, TS TP, TT Normales VWD
 Déficit de factores Vit K Dep. Hepatopatía TP, APTT TT, TS Normales V. Común, Déficit de factores Vit K Dep., Hepatopatía TP, APTT, TT, TS Hepatopatía, CID ↑ TP, APTT, TT TS Normal V. Final Fibrinógeno APTT TP, TT, TS Normal V. Intrínseca (FVIII, FIX, FXI, FXII) Hemofilia, VWD ↑ APTT, TS TP, TT Normales VWD.")
Presentaciones similares