Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Esclerosis Lateral Amiotrófica
AMA 2015 Bruno De Ambrosi FLENI

2
En forma clásica la esclerosis lateral amiotrófica se describía como una enfermedad que presentaba signos de motoneurona inferior (MNI) y superior (MNS), ya sea simultáneos o sucesivos, pero que finalmente sumados afectaban los tres/cuatro niveles en que “dividimos” el cuerpo (bulbar, cervical, torácico y lumbar).
 y superior (MNS), ya sea simultáneos o sucesivos, pero que finalmente sumados afectaban los tres/cuatro niveles en que dividimos el cuerpo (bulbar, cervical, torácico y lumbar).")
3
CLINICA Motoneurona superior:
- síndrome piramidal con hiperreflexia, espasticidad, clonus, Babinski, Hoffman, etc. Motoneurona inferior: - debilidad - atrofia - fasiculaciones.

4
Interrogantes Diagnóstico clínico. Confirmación.
Qué pruebas puedo realizar. Cómo transmitirlo. Tratamiento. Qué nos piden los pacientes. Qué les podemos ofrecer. Qué se puede sugerir. Hay forma de saber cuándo?

5
VARIANTES CLINICAS

6
Forma clásica tipo espinal:
- debilidad progresiva e indolora una extremidad - 60 años - signos tanto de MNS y MNI, rara la presencia de Babinski. - se extiend al resto del cuerpo afectando sectores contiguos.

7
Forma bulbar: - 20% - disfagia, disartria y defecto de la lengua - sobre vida menor - extensión a extremidades marcador de rápida evolución - 3-5% presenta inicio respiratorio prácticamente sin signos bulbares

8
Flail arm: - predominio masculino (4:1) - motoneurona inferior solo de miembros superiores en forma proximal - al segundo año expresan signos en otros segmentos, con una sobrevida mayor - Menos frecuente en miembros inferiores (“flail leg”, forma pseudo polineurítica, Marie-Patrikios o forma peroneal)
")
9
Atrofia muscular progresiva (AMP):
- Se compromete exclusivamente, al menos en el aspecto clínico, la MNI, diferenciándose del a atrofia músculo espinal del adulto por su asimetría, su compromiso proximal y/o distal y la rápida evolución.

10
Esclerosis lateral primaria (ELP):
- compromiso casi exclusivo de la MNS - progresión más lenta - 25% a los 4 años presentan signos de MNI. - sobrevida mayor, con un menor compromiso respiratorio y con menor pérdida de peso. - asimétrico, síndrome de Mills o hemiplejía progresiva, inicio pierna luego brazo homolateral

11
Compromiso extra motor
-Compromiso cognitivo: un 25% de los pacientes con ELA cumplen criterios para demencia fronto temporal (DFT), con un mayor compromiso conductual que cognitivo. Aún más frecuente es el defecto frontal o del lenguaje. ELA y DFT mismo espectro 50% de los pacientes con esta última tiene compromiso de motoneurona. -Compromiso extrapiramidal: la asociación ELA con parkinsonismo se refiere a pacientes con ELA y signos extrapiramidales no respondedores a la levodopa, expresándose principalmente como inestabilidad postural y caídas. Mucho menos frecuente es la asociación de ELA con enfermedad de Parkinson (EP) y se conoce como el síndrome de Brait-Fahn-Schwartz o complejo ELA- Parkinson. Suele expresarse la EP inicialmente de características habituales para años después sumarse la ELA. Si bien parecerían dos enfermedades aisladas, mutaciones de PARK7 puede generar EP, ELA y DFT.
, con un mayor compromiso conductual que cognitivo. Aún más frecuente es el defecto frontal o del lenguaje. ELA y DFT mismo espectro 50% de los pacientes con esta última tiene compromiso de motoneurona. -Compromiso extrapiramidal: la asociación ELA con parkinsonismo se refiere a pacientes con ELA y signos extrapiramidales no respondedores a la levodopa, expresándose principalmente como inestabilidad postural y caídas. Mucho menos frecuente es la asociación de ELA con enfermedad de Parkinson (EP) y se conoce como el síndrome de Brait-Fahn-Schwartz o complejo ELA- Parkinson. Suele expresarse la EP inicialmente de características habituales para años después sumarse la ELA. Si bien parecerían dos enfermedades aisladas, mutaciones de PARK7 puede generar EP, ELA y DFT.")
12
-Ataxia cerebelosa: muy infrecuente, puede asociarse con la variante C9orf72 y se ha descripto en algunos pacientes con ataxia espino cerebelosa (SCA). -Compromiso sensitivo: es frecuente en forma subjetiva y a veces es objetivo. La variante SOD1 d90a afecta el cordón posterior. -Compromiso autonómico: es muy rara la queja, pero en estudios se observan múltiples defectos subclínicos en varios componentes (gastrointestinal, sudoración, etc). En los pacientes con SOD1Asp90A y ELP son frecuentes los síntomas urinarios. -Ocular motor: es más frecuente en pacientes de larga sobrevida y con afectación frontal.
. En los pacientes con SOD1Asp90A y ELP son frecuentes los síntomas urinarios. -Ocular motor: es más frecuente en pacientes de larga sobrevida y con afectación frontal.")
13
DIAGNOSTICO

14
Las 3 i: idiota, intermedio, inteligente

15
Criterios diagnósticos
Presencia de: 1- Evidencia de degeneración de MNI según criterios clínicos, neurofisológicos o anátomo patológicos. 2- Evidencia de degeneración de MNS según criterios clínicos. 3- Progresiva diseminación de los signos o síntomas en la región o a otras, determinados por historia o examen; Ausencia de: 1- Evidencia clínica, electrofisiológica o patológica que pudiera explicar los signos de MNI y/o MNS. 2- Evidencia de neuroimágenes de otro proceso que pudiera explicar los hallazgos.

16
-ELA clínicamente definida: evidencia clínica o electrofisológica de lesión de MNI y MNS en región bulbar y dos espinales o en los tres niveles espinales. -ELA clínicamente probable: evidencia clínica de lesión de MNI y MNS en dos regiones con signos de MNS rostrales a los de MNI. -ELA clínicamente probable sustentada por laboratorio: signos clínicos de lesión de MNS y MNI en una región o signos de MNS en una sola región, con signos definidos por electromiografía de compromiso de MNI en al menos dos regiones. -ELA clínicamente posible: signos clínicos de lesión de MNS y MNI en una sola región o los signos de MNS se encuentran en dos o más regiones, o los signos de MNI son rostrales a los MNS.

17
Electrofisiología Puede preceder a la expresión clínica y debe ser amplia. En reposo la actividad espontánea, que representa la denervación activa, suele ser intensa y se caracteriza por la presencia de fibrilaciones, ondas agudas positivas y fasciculaciones Ante la actividad voluntaria encontramos los signos de reinervación: PUM de duración y amplitud aumentada Las velocidades de conducción motoras suelen ser normales inicialmente para reducir su amplitud y en menor medida su velocidad como representación de la pérdida axonal y la atrofia muscular. Las velocidades sensitivas suelen preservarse. La onda F se afecta principalmente en su dispersión y frecuencia, con relativa preservación de la latencia. La estimulación repetitiva de baja frecuencia puede dar decremento sobre todo en músculos distales. Potenciales motores

18
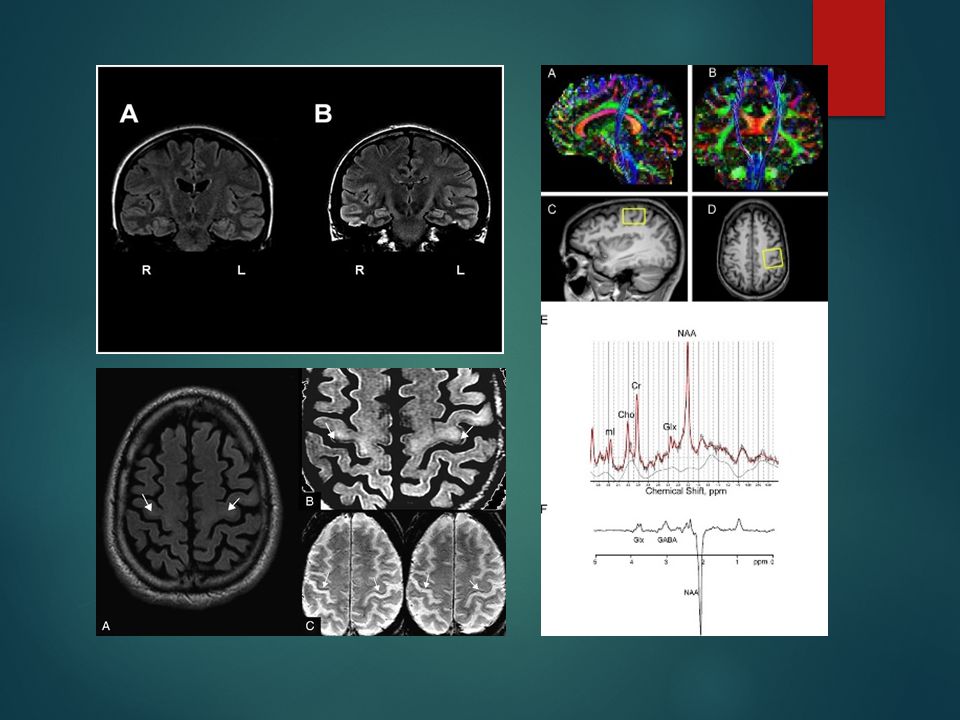
Neuroimágenes RMN convencional: se pueden observar
- alteraciones en la intensidad el tracto cortico espinal en T1, T2 y FLAIR. - línea hipodensa en el giro precentral en T2 y FLAIR, hallazgo que puede observarse en gente sana o en otras enfermedades neurodegenerativas. Su hallazgo en las secuencias de densidad protónica, parecerían ser mucho más específicas para el diagnóstico de ELA. - atrofia del giro precentral en pacientes con ELA y ELP. - morfometría basada en voxel disminución del volumen cortical en regiones extra motoras aún sin franca demencia.

19
Tensor de difusión (DTI): defectos en el haz cortico espinal, principalmente en el brazo posterior de la cápsula, aunque también hay gran cantidad de trabajos que sostiene la utilidad de esta técnica para valorar severidad y progresión de la enfermedad así como el compromiso de múltiples vías no motoras.
: defectos en el haz cortico espinal, principalmente en el brazo posterior de la cápsula, aunque también hay gran cantidad de trabajos que sostiene la utilidad de esta técnica para valorar severidad y progresión de la enfermedad así como el compromiso de múltiples vías no motoras.")
20
Espectroscopía: - disminución en la relación N acetil aspartato (NAA) con la creatina - modesta correlación entre concentración de NAA y sus relaciones y las manifestaciones clínicas - aumento del mioinositol como expresión de una mayor actividad de la células gliales.

22
Genética 5-10% de los cuadros son familiares (ELAf)
Se ha podido identificar el defecto genético en el 60% de los casos. 40% corresponde a un defecto del cromosoma 9 (C9ORF72), el 20% al CuZn superóxido dismutasa (SOD1), y un 5% y 3% para FUS y TARDBP respectivamente. Pese a la ausencia de historia familiar, aproximadamente el 10% de los pacientes tiene las mutaciones de los cuadros definidos como familiares, lo que hace difuso el límite de esta división.
, el 20% al CuZn superóxido dismutasa (SOD1), y un 5% y 3% para FUS y TARDBP respectivamente. Pese a la ausencia de historia familiar, aproximadamente el 10% de los pacientes tiene las mutaciones de los cuadros definidos como familiares, lo que hace difuso el límite de esta división.")
23
TRATAMIENTO

24
Tratamiento Habitualmente se indica terapia física y ocupacional
Plan de trabajo físico adaptado a las necesidades y posibilidades del paciente Premisa principal evitar el esfuerzo intenso y el agotamiento. Supeditada a la experiencia de los profesionales participantes, dada la carencia de guías definitivas sobre este tema evaluación permanente e indicación de material ortésico y de adaptación, con el objetivo principal de mantener la independencia y seguridad del paciente

25
Fármacos -Sialorrea: extremadamente frecuente. La amitripilitina en dosis bajas. Opciones de uso local son la atropina, el glicopirrolato y la escopolamina. Infiltración de parótidas con toxina botulínica y se ha descripto la irradiación de las mismas. -Secreciones bronquiales: podrían beneficiarse de mucolíticos como N acetilcisteína, nebulizaciones con agonistas beta, técnicas de asistencia manual de la tos, el uso de aparatos de insuflación mecánica así como aspiradores. -Labilidad emocional seudo bulbar: se utiliza amitriptilina así como IRSS y la combinación de quinidina con dextrometorfano. -Calambres: magnesio o quinidina habiéndose utilizado también anti epilépticos. fisioterapia o hidroterapia.

26
Fármacos - Espasticidad: actividad física frecuente, medicación como baclofeno o tizanidina. bomba de baclofeno intratecal. -Depresión y ansiedad: de utiliza habitualmente amitriptilina e IRSS, dependiendo en general de otros síntomas acompañantes. También es posible usar benzodiacepinas. -Fatiga y debilidad: puede ser útil la creatina, así como también se ha utilizado modafinilo y anticolinesterásicos pero estos últimos pueden aumentar las fasciculaciones. -Dolor: frecuente en estadios avanzados de la enfermedad, juega un rol fundamental la terapia física así como el uso racional de los distintos analgésicos.

27
Evaluación y manejo respiratorio
Evaluación: capacidad vital forzada (CVF) y la presión inspiratoria máxima (Pimax) y la presión espiratoria máxima (Pemax). Algunos parámetros para considerar el uso de ventilación no invasiva serían la ortopnea, somnolencia diurna con una Pimax menor al 60% previsto, hipercapnia diurna, desaturación de oxígeno diurna o un índice de apneas mayor a 10, CVF menor a 50% con o sin síntomas respiratorios. El uso de la ventilación no invasiva (VNI): combinación entre los síntomas y la evidencia diagnóstica de debilidad respiratoria. BiPAP (bilevel positive airway pressure) es el dispositivo más utilizado.. Equipos de insuflación y exuflación mecánica en pacientes con pico flujo disminuido y con defecto en el manejo de secreciones. Otra opción válida el uso de “breath stacking” con asistencia manual de la tos. Traqueostomía y ventilación mecánica: si bien no es claro cuál es el mejor momento para plantear su realización, debe considerarse en pacientes que no toleran la VNI con CVF menor a 50% o disnea o en los que ya resulta insuficiente la misma pese a su optimización y en general esto se asocia con un pobre manejo de las secreciones. En general, es razonable su discusión antes que el paciente se encuentre en situación de “necesidad” y tener en claro los deseos del mismo. La sobrevida luego de la misma va de 12 a 36 meses y en general la causa de muerte es una infección del tracto respiratorio. Se estima que más del 80% de los pacientes traqueostomizados están conformes con la decisión tomada y la mitad de ellos.
 y la presión inspiratoria máxima (Pimax) y la presión espiratoria máxima (Pemax). Algunos parámetros para considerar el uso de ventilación no invasiva serían la ortopnea, somnolencia diurna con una Pimax menor al 60% previsto, hipercapnia diurna, desaturación de oxígeno diurna o un índice de apneas mayor a 10, CVF menor a 50% con o sin síntomas respiratorios. El uso de la ventilación no invasiva (VNI): combinación entre los síntomas y la evidencia diagnóstica de debilidad respiratoria. BiPAP (bilevel positive airway pressure) es el dispositivo más utilizado.. Equipos de insuflación y exuflación mecánica en pacientes con pico flujo disminuido y con defecto en el manejo de secreciones. Otra opción válida el uso de breath stacking con asistencia manual de la tos. Traqueostomía y ventilación mecánica: si bien no es claro cuál es el mejor momento para plantear su realización, debe considerarse en pacientes que no toleran la VNI con CVF menor a 50% o disnea o en los que ya resulta insuficiente la misma pese a su optimización y en general esto se asocia con un pobre manejo de las secreciones. En general, es razonable su discusión antes que el paciente se encuentre en situación de necesidad y tener en claro los deseos del mismo. La sobrevida luego de la misma va de 12 a 36 meses y en general la causa de muerte es una infección del tracto respiratorio. Se estima que más del 80% de los pacientes traqueostomizados están conformes con la decisión tomada y la mitad de ellos.")
28
Evaluación y manejo de la deglución
Tos durante o luego de deglutir, dificultad en masticación, babeo, restos de comida, cambios en la voz luego de deglutir, odinofagia o pérdida de peso. Queja por sialorrea en parte secundario al defecto del control motor de la deglución, agregándose a esto saliva más espesa. Estadíos tempranos principalmente defectos orales y mínimos faríngeos: cambios de hábitos mínimos para optimizar deglución y evitar una ingesta ya sea sólida o líquida insuficiente. Posteriormente es necesario modificar las consistencias lo que requiere dieta blanda, uso de líquidos espesados, evitar texturas diferentes simultáneas, estimulación sensorial, etc. Cuando no puede cubrir sus requerimientos dietarios se recomienda la gastrostomía que no implica necesariamente la suspensión de la ingesta oral. Más avanzado ante la incapacidad de manejar alimentos y secreciones se recomienda suspender la vía oral.

Presentaciones similares




 Autora: Lic. Yarelis Castellanos Vargas E mail: ycastellanos@neuro.ciren.cu.>")



 Novedades>")










