Descargar la presentación
La descarga está en progreso. Por favor, espere

1
I-Alteraciones Estructurales
Química Biológica Patológica HEMOGLOBINOPATIAS I-Alteraciones Estructurales Tema:3 (3) Dra. Silvia Varas
 Dra. Silvia Varas.")
2
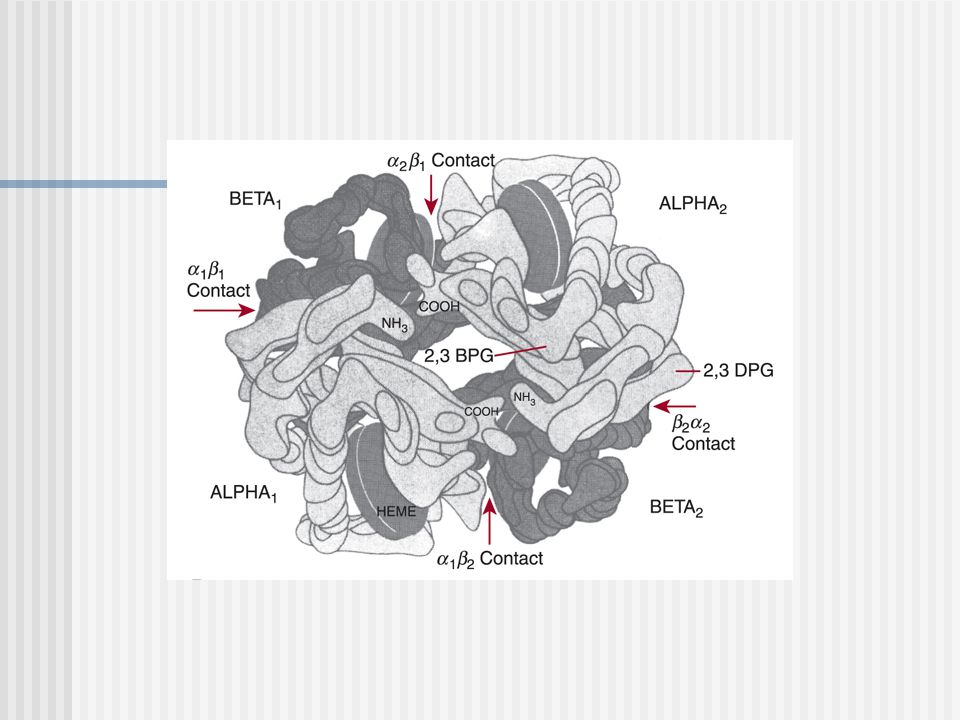
Esquema de la Molécula de Hemoglobina

3
Ontogenia cadenas de globina

4
1

5
, , , ,

6
Síntesis de cadenas de globina
% en embrión, feto y lactante

7
UNION DEL HEM A LA GLOBINA Y AL OXÍGENO
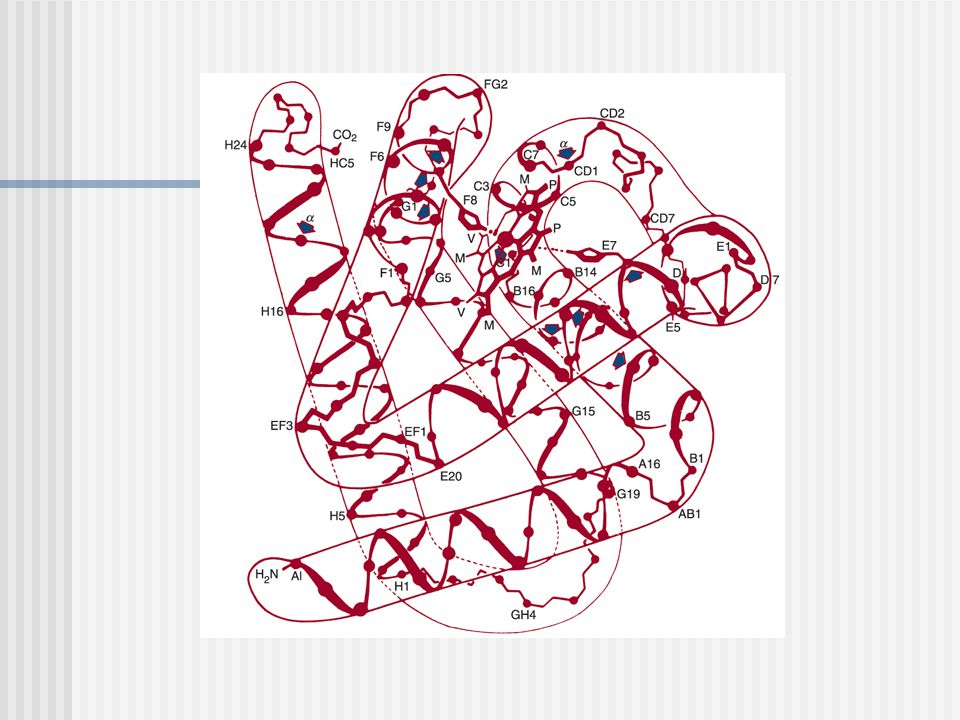
El hierro se une la Hys F8 ( histidina proximal) Al otro lado se encuentra la Hys E7 (histidina distal) donde se une el O2, CO2 y CO Cada subunidad esta constituida por 8 hélices rotuladas AH conectadas por uniones polipeptídicas cortas
 Al otro lado se encuentra la Hys E7 (histidina distal) donde se une el O2, CO2 y CO. Cada subunidad esta constituida por 8 hélices rotuladas AH conectadas por uniones polipeptídicas cortas.")
8
RESIDUOS EN CONTACTO CON EL HEM
Los residuos no polares en los segmentos C, CD, E, F y FG protegen la abertura al impedir que el agua penetre.

9
BOLSILLO DEL HEM La jaula hidrofóbica donde se encuentra el hem es la fuerza estabilizadora principal para la unión del hem a la proteína El medio no polar dificulta la oxidación de Fe2+ a Fe3+.

10
Desoxihemoglobina Oxihemoglobina
La oxigenación pone a las cadenas mas juntas Oxihemoglobina

11
Estado Desoxihb o Tenso (T)
Estado (T): hay cerca 22 contactos (12 puentes de hidrogeno) Estado Oxihb o Relajado (R) Estado R: hay cerca 40 contactos (19 puentes de hidrogeno)
: hay cerca 22 contactos (12 puentes de hidrogeno) Estado Oxihb o Relajado (R) Estado R: hay cerca 40 contactos (19 puentes de hidrogeno)")
12
La mayoría de los movimientos de oxigenación/deoxigenación toma lugar en las interfaces 12 (21)
Las interfaces 11 (22) permanecen inmóviles Desoxigenación: Oxigenación:
 permanecen inmóviles. Desoxigenación: Oxigenación:")
13
HEMOGLOBINOPATÍAS ALTERACIONES CUALITATIVAS ALTERACIONES CUANTITATIVAS
Hemoglobinopatías estructurales ALTERACIONES CUANTITATIVAS Síndromes Talasémicos

14
Clasificación: ESTRUCTURALES Cadenas Cadenas Cadenas Cadenas
Fusión de Cadenas: TALASEMIAS - talasemia - talasemia - talasemia - talasemia - talasemia PERSISTENCIA HEREDITARIA DE HEMOGLOBINA F Con deleción Sin deleción Ligado al cluster No Ligado al cluster

15
Clasificación según Fenotipo
I-Propiedades físico-químicas alteradas A-HbS (polimerización deoxihb S): síndromes drepanocíticos B-HbC (cristalización): anemia hemolítica, microcitosis II-Variantes de Hemoglobina Inestables A-Anemia hemolítica de cuerpos de Heinz Congénita III- Variantes con afinidad al O2 alterada A-Variantes con alta afinidad: eritrocitosis B-Variantes con baja afinidad: anemia, cianosis IV-Hemoglobina M A-Metahemoglobinemia, cianosis. V- Variantes que causan un fenotipo talasémico
: síndromes drepanocíticos. B-HbC (cristalización): anemia hemolítica, microcitosis. II-Variantes de Hemoglobina Inestables. A-Anemia hemolítica de cuerpos de Heinz Congénita. III- Variantes con afinidad al O2 alterada. A-Variantes con alta afinidad: eritrocitosis. B-Variantes con baja afinidad: anemia, cianosis. IV-Hemoglobina M A-Metahemoglobinemia, cianosis. V- Variantes que causan un fenotipo talasémico.")
16
VARIANTES ESTRUCTURALES DE HEMOGLOBINAS
NOMENCLATURA PATOLOGIA MOLECULAR: a-Sustituciones de una única base. HbS b-Variantes de cadena de hemoglobina elongadas (cadenas alargadas). Hb CS c-Cadenas de globinas truncadas (deleción de fragmentos). Hb Leiden, Gun Hill d- Hemoglobinas de fusión (hibridización anómala). Hb Lepore
. Hb CS. c-Cadenas de globinas truncadas (deleción de fragmentos). Hb Leiden, Gun Hill. d- Hemoglobinas de fusión (hibridización anómala). Hb Lepore.")
17
a-Sustituciones de una única base.
b-Variantes de cadena de Hb elongadas.

18
Mutantes de Terminación: Cadenas Alargadas
Los mutantes de terminación son todos variantes de la cadena alfa globina donde hay una única sustitución de base en el codón stop UAA .

19
Hb Constant Spring. 1 y 2 Adulto Normal; 3 y 4 heterocigota para HbCS y º-talasemia con HbH;5:Adulto Normal; y 6 componente heterocigota para º-talasemia y HbCS

20
c-Cadenas de globinas truncadas
En este caso, un o más aminoácidos adyacentes se pierden, pero se conserva el resto de la cadena normal. Estas variantes involucran la deleción de 1 o más codones intactos, no afectando el marco de lectura de la proteína restante. Consisten en una perdida de uno o varios nucleótidos en la secuencia del DNA que codifica la síntesis de una cadena de globina. Son ejemplos de deleciones las Hb Freiburg (deleción de un aminoácido), Hb Lyon (deleción de dos aminoácidos) y Hb GunHill (deleción de cinco aminoácidos).
, Hb Lyon (deleción de dos aminoácidos) y Hb GunHill (deleción de cinco aminoácidos).")
21
d- Hemoglobinas de fusión Hemoglobina Lepore Hemoglobina Kenya
-El gen híbrido δβ se llama Lepore y El gen híbrido βδ se llama anti-Lepore Hb Lepore tiene los primeros 20 a 80 aminoácidos de las cadenas y los últimos 50 a 100 aminoácidos del extremo C-terminal de la cadena .

22
Clasificación según Fenotipo
I-Propiedades físico-químicas alteradas A-HbS (polimerización deoxihb S): síndromes drepanocíticos B-HbC (cristalización): anemia hemolítica, microcitosis II-Variantes de Hemoglobina Inestables A-Anemia hemolítica de cuerpos de Heinz Congénita III- Variantes con afinidad al O2 alterada A-Variantes con alta afinidad: eritrocitosis B-Variantes con baja afinidad: anemia, cianosis IV-Hemoglobina M A-Metahemoglobinemia, cianosis. V- Variantes que causan un fenotipo talasémico
: síndromes drepanocíticos. B-HbC (cristalización): anemia hemolítica, microcitosis. II-Variantes de Hemoglobina Inestables. A-Anemia hemolítica de cuerpos de Heinz Congénita. III- Variantes con afinidad al O2 alterada. A-Variantes con alta afinidad: eritrocitosis. B-Variantes con baja afinidad: anemia, cianosis. IV-Hemoglobina M A-Metahemoglobinemia, cianosis. V- Variantes que causan un fenotipo talasémico.")
23
I-Hemoglobina con Propiedades Fco-Qco Alteradas
Hemoglobina S Hemoglobina C Hemoglobina E Hemoglobina D

24
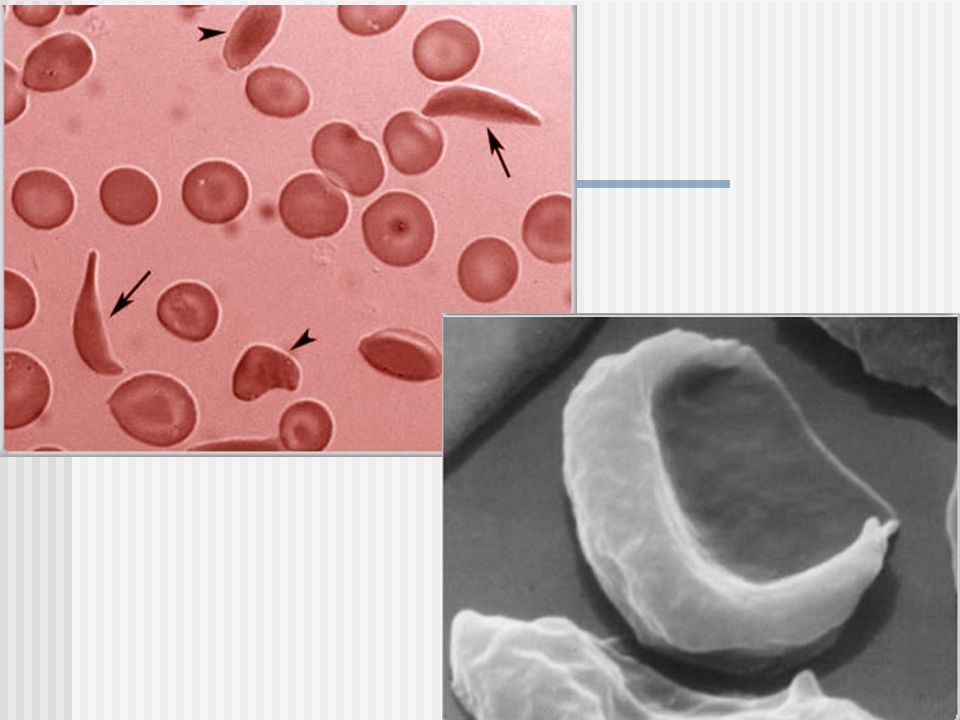
Anemia Falciforme, Drepanocítica, en Forma de Hoz (sickle), o HbS
6 Cadena

25
GTGGAG WT: Ac.Glutámico

26
GTGGAG S: Valina

28
Fisiopatología: POLIMERIZACION DE HEMOGLOBINA
La presencia de HbS, permite a esta polimerizar cuando se desoxigena (Estado T), ya que la valina (pero NO Ac.Glutámico) puede unirse a ésteres complementarios en cadenas de globina adyacentes
, ya que la valina (pero NO Ac.Glutámico) puede unirse a ésteres complementarios en cadenas de globina adyacentes.")
29
2 La valina (2) se une a un bolsillo hidrófobo sobre la subunidad 1 de una molécula vecina de oxihemoglobina.
 se une a un bolsillo hidrófobo sobre la subunidad 1 de una molécula vecina de oxihemoglobina.")
30
a b c c/polímero= 14 haces longitudinales= cuerpo tactoide
Estructura cilíndrica, insoluble y rígida c Micrografía electrónica de fibras teñidas negativamente de HbS y la estructura deducida por reconstrucción de la imagen tridimensional. Las fibras reconstruidas esta presente como modelos de balones, (cada balón representa un tetrámero de HbS). Los modelos se presentan como la cubierta exterior (a), el núcleo interno (b) y una combinación de ambos filamentos el externo y el interno (c)
. Los modelos se presentan como la cubierta exterior (a), el núcleo interno (b) y una combinación de ambos filamentos el externo y el interno (c)")
31
Fibras de desoxihemoglobina S

32
Fisiopatología: POLIMERIZACION DE HEMOGLOBINA DESHIDRATACION CELULAR
DISMINUCIÓN DEL NO Y DISFUNCIÓN DE ENDOTELIO ADHESIVIDAD CELULAR ANORMAL INFLAMACION ISQUEMIA-REPERFUSIÓN LA ACTIVACIÓN DEL SISTEMA DE COAGULACIÓN VASCULOPATIA CRONICA

33
6º 2º 1º 4º 5º 3º 7º VCAM (vascular cell adhesion molecule )
")
34
SCAVENGING o BARRIDO DE OXIDO NITRICO (NO)
")
35
HEMOLISIS INTRAVASCULAR
SCAVENGING de NO Y DISFUNCIÓN DE ENDOTELIO HEMOLISIS INTRAVASCULAR

36
Diagnóstico Anemia con HbS:

37
Características clínicas
1. Fase estacionaria. Corresponde, generalmente, a los primeros años de vida (1-4 años), y sus manifestaciones clínicas son las propias de un síndrome hemolítico crónico moderado o intenso (anemia, palidez cutáneo-mucosa, subictericia conjuntival y retraso del crecimiento óseo y gonadal). 2. Fase de expresividad aguda. Se inicia a partir de los 4 años de edad, con agravamiento del cuadro anémico (Hb < 80 g/L) y aparición de diversas manifestaciones clínicas de carácter agudo debidas a las crisis vasooclusivas que afectan de forma importante a diversos órganos, aunque muy especialmente al pulmón, al riñón y al tejido óseo (drepanocitosis). Las crisis vasooclusivas constituyen, de hecho, la manifestación clínica más característica y grave de la anemia falciforme y, muchas veces, el primer síntoma. Se trata de ataques, muy dolorosos y pasajeros, que pueden durar días o semanas. 3. Fase de expresividad crónica. Es propia de los pacientes que han logrado sobrevivir la primera infancia, por lo que es característica de la adolescencia y la edad adulta. El carácter evolutivo crónico de la anemia falciforme afecta de forma importante al crecimiento y desarrollo corporal, al sistema nervioso central, cardiovascular, pulmonar, hepatobiliar y gastrointestinal. Asimismo, condiciona lesiones graves de la función renal y trastornos visuales que pueden conducir a la ceguera
, y sus manifestaciones clínicas son las propias de un síndrome hemolítico crónico moderado o intenso (anemia, palidez cutáneo-mucosa, subictericia conjuntival y retraso del crecimiento óseo y gonadal). 2. Fase de expresividad aguda. Se inicia a partir de los 4 años de edad, con agravamiento del cuadro anémico (Hb < 80 g/L) y aparición de diversas manifestaciones clínicas de carácter agudo debidas a las crisis vasooclusivas que afectan de forma importante a diversos órganos, aunque muy especialmente al pulmón, al riñón y al tejido óseo (drepanocitosis). Las crisis vasooclusivas constituyen, de hecho, la manifestación clínica más característica y grave de la anemia falciforme y, muchas veces, el primer síntoma. Se trata de ataques, muy dolorosos y pasajeros, que pueden durar días o semanas. 3. Fase de expresividad crónica. Es propia de los pacientes que han logrado sobrevivir la primera infancia, por lo que es característica de la adolescencia y la edad adulta. El carácter evolutivo crónico de la anemia falciforme afecta de forma importante al crecimiento y desarrollo corporal, al sistema nervioso central, cardiovascular, pulmonar, hepatobiliar y gastrointestinal. Asimismo, condiciona lesiones graves de la función renal y trastornos visuales que pueden conducir a la ceguera.")
39
Otras variantes estructurales de Hb
Hemoglobina C: fue la segunda variante a ser identificada electroforeticamente. Resulta de la sustitución de Lisina por Ac.Glutámico en la posición 6 de la cadena de de globina. Existe un número de observaciones que sugieren que la Hemoglobina C es menos soluble que la Hemoglobina A y tiene tendencia a cristalizar en el eritrocito. Hemoglobina D: este termino se usa par describir un numero de variantes de hemoglobinas que tienen una velocidad de migración idéntica a HbS en una electroforesis a pH alcalino. La mas común de estas hemoglobinas es la Hb DLOS ANGELES, sus eritrocitos son normales excepto por el elevado numero de células target. Hemoglobina E: Es la variante más común en el mundo. Resulta de la sustitución de Acido Glutámico por Lisina en la posición 26 en la cadena de globina.

40
Frotis de sangre. Enfermedad Hb SS con sickle- cells.
Microscopia contraste de fase. Note 3 sickled cells con proyecciones (formación tactoide). Frotis de sangre. Enfermedad Hb SS con sickle- cells. Frotis de sangre de enfermedad con Hb CC. Numerosas target cells y una población de microesferocitos (hipercromática).De las células no esferociticas, virtualmete todas son target cells. Frotis de sangre de Enfermedad Hb SC. Note frecuencia de target cells, característica de Hb C y las células son pequeñas y densas, irregular, refleja su contenido de Hb S.
. Frotis de sangre. Enfermedad Hb SS con sickle- cells. Frotis de sangre de enfermedad con Hb CC. Numerosas target cells y una población de microesferocitos (hipercromática).De las células no esferociticas, virtualmete todas son target cells. Frotis de sangre de Enfermedad Hb SC. Note frecuencia de target cells, característica de Hb C y las células son pequeñas y densas, irregular, refleja su contenido de Hb S.")
41
FS de enfermedad Hb EE. Hipocromia, anisocitosis y target cells.
FS de enfermedad con Hb C postesplenectomia. Note las inclusiones semejantes a una barra en dos células como resultado de cristalización de Hb C. Estas células son virtualmente removidas en pac. con bazo. Enfermedad de Hb CC postesplenectomia. Microscopia de contraste de Fase. Note la barra cristalina de Hb C in una célula. FS de enfermedad Hb DD. Note target cells en una población de esferocitas pequeños, poikilocitos, y pequeños fragmentos de GR. FS de enfermedad Hb EE. Hipocromia, anisocitosis y target cells.

42
Clasificación según Fenotipo
I-Propiedades físico-químicas alteradas A-HbS (polimerización deoxiHb S): síndromes drepanocíticos B-HbC (cristalización): anemia hemolítica, microcitosis II-Variantes de Hemoglobina Inestables A-Anemia hemolítica de cuerpos de Heinz Congénita III- Variantes con afinidad al O2 alterada A-Variantes con alta afinidad: eritrocitosis B-Variantes con baja afinidad: anemia, cianosis IV-Hemoglobina M A-Metahemoglobinemia, cianosis. V- Variantes que causan un fenotipo talasémico
: síndromes drepanocíticos. B-HbC (cristalización): anemia hemolítica, microcitosis. II-Variantes de Hemoglobina Inestables. A-Anemia hemolítica de cuerpos de Heinz Congénita. III- Variantes con afinidad al O2 alterada. A-Variantes con alta afinidad: eritrocitosis. B-Variantes con baja afinidad: anemia, cianosis. IV-Hemoglobina M. A-Metahemoglobinemia, cianosis. V- Variantes que causan un fenotipo talasémico.")
44
II-Desordenes de Hemoglobinas Inestables
Anemia Hemolítica de Cuerpos de Heinz Congénitos, (CHBA) (Congenital Heinz body hemolitic anemia) Tipo 1: Sustituciones en la vecindad del bolsillo del hem . La unión del hem a la globina involucra interacciones específicas con residuos de aminoácido no polares en las regiones CD, E, F, y FG
 (Congenital Heinz body hemolitic anemia) Tipo 1: Sustituciones en la vecindad del bolsillo del hem . La unión del hem a la globina involucra interacciones específicas con residuos de aminoácido no polares en las regiones CD, E, F, y FG.")
45
Desordenes de Hemoglobinas Inestables
Anemia Hemolítica de Cuerpos de Heinz Congénitos, (CHBA) (Congenital Heinz body hemolitic anemia) Tipo 2: Ruptura Estructura Secundaria de -hélice. Prolina no puede participar excepto como parte de uno de los tres residuos iniciales. Hay sustituciones de Pro Tipo 3: Mutaciones en la interfaz 11 Tipo 4 : Deleciones de aminoácidos Tipo 5: Cadenas Elongadas
 (Congenital Heinz body hemolitic anemia) Tipo 2: Ruptura Estructura Secundaria de -hélice. Prolina no puede participar excepto como parte de uno de los tres residuos iniciales. Hay sustituciones de Pro. Tipo 3: Mutaciones en la interfaz 11. Tipo 4 : Deleciones de aminoácidos. Tipo 5: Cadenas Elongadas.")
46
Formación Cuerpos Heinz
OxiHb (6HS) Formación Cuerpos Heinz NADH NAD+ - Superóxido dismutasa MetaHb (6HS) O2 Hb sin hem Hemicromo 1 (Reversible) Hemicromo 2 (Irreversible) R H2O2 GSH Catalasa Cadenas precipitadas Precipita (4 HS) I Glutation peroxidasa GSSG Cuerpos de Heinz (unido MP) H2O LISIS
 Formación Cuerpos Heinz. NADH. NAD+ - Superóxido dismutasa. MetaHb (6HS) + O2. Hb sin hem. Hemicromo 1 (Reversible) Hemicromo 2 (Irreversible) R. H2O2. GSH. Catalasa. Cadenas precipitadas. Precipita (4 HS) I. Glutation peroxidasa. GSSG. Cuerpos de Heinz (unido MP) H2O. LISIS.")
47
Mecanismo de formación de los Cuerpos de Heinz
Autoxidación ión superóxido Los hemicromos son moléculas de metahemoglobina en las que la quinta o sexta posición de coordinación con el hierro se une en otra posición de la normal, generando una distorsión de la molécula

48
Mecanismo de Hemólisis:
Una teoría propuesta como mecanismo de hemólisis seria la siguiente (1º) un aumento de la oxidación y fosforilación de tirosina en la proteína AE1; unión de los hemicromos a la proteína Banda 3 (2º) reclutamiento progresivo de AE1 fosforilada en grandes complejos en membrana lo cuales contienen hemicromos y (3º) paralela lisis de los eritrocitos y una masiva liberación de vesículas que contienen hemicromos. AE1, anion exchanger1 =Banda3.
 un aumento de la oxidación y fosforilación de tirosina en la proteína AE1; unión de los hemicromos a la proteína Banda 3. (2º) reclutamiento progresivo de AE1 fosforilada en grandes complejos en membrana lo cuales contienen hemicromos y. (3º) paralela lisis de los eritrocitos y una masiva liberación de vesículas que contienen hemicromos. AE1, anion exchanger1 =Banda3.")
49
II-Mecanismos de Remoción Fagocítica de Eritrocitos
La secuencia de eventos sería: a- Unión de los hemicromos a la proteína Banda 3, b- Oxidación de los dominios citoplasmático de Banda 3, c- Generación de agregados de alto peso molecular de Banda 3 (clustering de banda 3) que une IgG y complemento, d- Opsonización con Ac anti- Banda3 e- Reconocimiento por parte del sistema mononuclear-fagocítico y fagocitosis por macrófagos.
 que une IgG y complemento, d- Opsonización con Ac anti- Banda3. e- Reconocimiento por parte del sistema mononuclear-fagocítico y fagocitosis por macrófagos.")
50
Clasificación según Fenotipo
I-Propiedades físico-químicas alteradas A-HbS (polimerización deoxiHb S): síndromes drepanocíticos B-HbC (cristalización): anemia hemolítica, microcitosis II-Variantes de Hemoglobina Inestables A-Anemia hemolítica de cuerpos de Heinz Congénita III- Variantes con afinidad al O2 alterada A-Variantes con alta afinidad: eritrocitosis B-Variantes con baja afinidad: anemia, cianosis IV-Hemoglobina M A-Metahemoglobinemia, cianosis. V- Variantes que causan un fenotipo talasémico
: síndromes drepanocíticos. B-HbC (cristalización): anemia hemolítica, microcitosis. II-Variantes de Hemoglobina Inestables. A-Anemia hemolítica de cuerpos de Heinz Congénita. III- Variantes con afinidad al O2 alterada. A-Variantes con alta afinidad: eritrocitosis. B-Variantes con baja afinidad: anemia, cianosis. IV-Hemoglobina M A-Metahemoglobinemia, cianosis. V- Variantes que causan un fenotipo talasémico.")
52
III-

53
Clasificación según Fenotipo
I-Propiedades físico-químicas alteradas A-HbS (polimerización deoxiHb S): síndromes drepanocíticos B-HbC (cristalización): anemia hemolítica, microcitosis II-Variantes de Hemoglobina Inestables A-Anemia hemolítica de cuerpos de Heinz Congénita III- Variantes con afinidad al O2 alterada A-Variantes con alta afinidad: eritrocitosis B-Variantes con baja afinidad: anemia, cianosis IV-Hemoglobina M A-Metahemoglobinemia, cianosis. V- Variantes que causan un fenotipo talasémico
: síndromes drepanocíticos. B-HbC (cristalización): anemia hemolítica, microcitosis. II-Variantes de Hemoglobina Inestables. A-Anemia hemolítica de cuerpos de Heinz Congénita. III- Variantes con afinidad al O2 alterada. A-Variantes con alta afinidad: eritrocitosis. B-Variantes con baja afinidad: anemia, cianosis. IV-Hemoglobina M A-Metahemoglobinemia, cianosis. V- Variantes que causan un fenotipo talasémico.")
54
TALASEMIAS - talasemia - talasemia - talasemia - talasemia
PERSISTENCIA HEREDITARIA DE HEMOGLOBINA F Con deleción Sin deleción Ligado al cluster No Ligado al cluster

55
Alguna pregunta?

56
Vamos a la Talasemias

Presentaciones similares