Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Glóbulos blancos. Leucopenias. Trastornos de los GB.
Fisiopatología Glóbulos blancos. Leucopenias. Trastornos de los GB. Ana Martínez & Natalia Portillo.

2
Glóbulos blancos. Son células sanguíneas. También reciben el nombre de Leucocitos o células blancas. Leuco (Leuko): Prefijo que significa Blanco. Cito (Cyte): Sufijo que significa Célula Su concentración en el adulto normal es de – mm3 de sangre. Se forman en parte en la médula ósea y en parte en el tejido linfático. En un frotis los leucocitos tienen forma esférica, en el tejido son pleomórficos.
: Prefijo que significa Blanco. Cito (Cyte): Sufijo que significa Célula. Su concentración en el adulto normal es de – mm3 de sangre. Se forman en parte en la médula ósea y en parte en el tejido linfático. En un frotis los leucocitos tienen forma esférica, en el tejido son pleomórficos.")
3
Clasificación de los GB.
Neutrófilos. Granulares Eosinófilos. Basófilos. Monocitos. Agranulares Linfocitos.

4
Médula Ósea y Hematopoyesis

5
Neutrófilos. Mide 9 a 12 um Representa un 62% del total de leucocitos.
Núcleo segmentado conectados por filamentos. Mide 9 a 12 um Representa un 62% del total de leucocitos. Tiene gránulos inespecíficos que no reaccionan ni al colorante ácido, ni al básico. Son la primera línea de defensa frente a la invasión por bacterias.

6
Eosinófilos. Núcleo bilobulado con distintos gránulos citoplasmáticos.
Representan el 2,3 % del total de leucocitos. Los gránulos inespecíficos fijan el colorante ácido.

7
Basófilos Núcleos irregulares. En forma de “S”
Gránulos que fijan el colorante básico. Miden 10um de diámetro. Los gránulos específicos son vesículas secretorias que contienen Histamina, Heparina, Bradicina y serotonina Representan el 0,4 % del total.

8
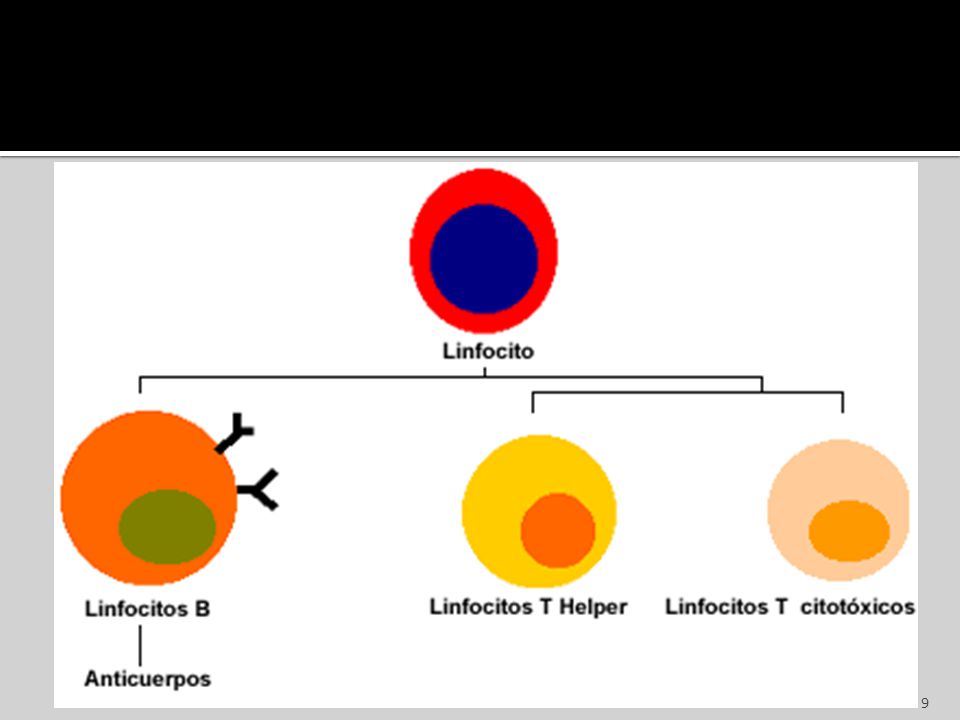
Linfocitos Representan un 30% del total.
Núcleo esférico, muy grande, de escaso citoplasma. No contienen ningun granulo identificable en su citoplasma 8 a 10 um de diametro Se clasifican según el “CD”, son de dos tipos. 85% corresponden a las células T, 15% a las células B.

10
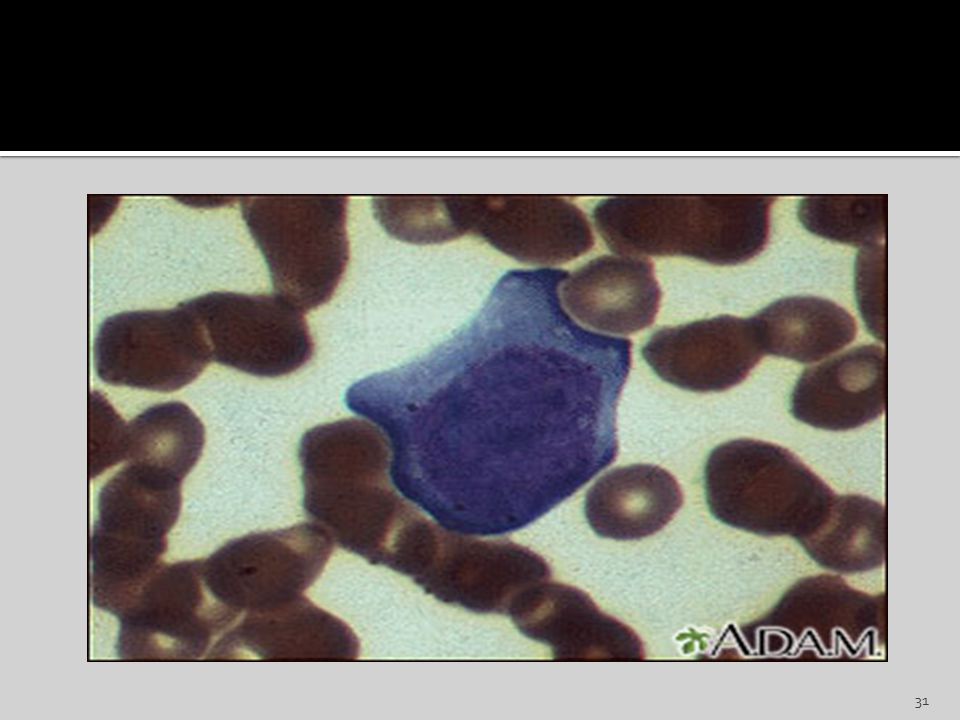
Monocito Son los glóbulos blancos más grandes.
Llegan a medir hasta 18 um de diámetro. Constituyen entre el 3 y 8% del total de leucocitos. Presenta núcleo en general arriñonado, lobulado, que se tiñe irregularmente violeta-azulado. Cuando ingresan a los tejidos, son macrófagos .

11
Trastornos de los glóbulos blancos..

12
T. No neoplásicos de los glóbulos blancos:
- Leucocitosis: Linfadenitis Aguda o Crónica - Leucopenias: Neutropenia. Mononucleosis Infecciosa. T. Neoplásicos de origen Hematopoyético y linfoide: - Linfomas: Enf. De Hodgkin y no Hodgkin. - Leucemias: Aguda, crónicas - Discrasia de los plasmocitos: mieloma múltiple

13
Leucocitosis.. Es el aumento en el número de células blancas de la sangre. Decimos que hay leucocitosis cuando la cifras de GB es superior a por mm³ de sangre. Es una reacción que se observa habitualmente en diversos procesos inflamatorios. El número de leucocitos que contiene la sangre periférica depende de varios factores, entre ellos: La magnitud de los fondos comunes de los precursores mieloides y linfoides, y del fondo común de las células de reserva. La velocidad con que las células pasan desde el fondo común de reserva a la circulación. El porcentaje de células que experimenta la marginación en cualquier momento. La velocidad con que las células se extravasan pasando desde la sangre periférica a los tejidos.

14
La homeostasis de los leucocitos se mantiene gracias a las citocinas, factores de crecimiento y moléculas de adhesión, que intervienen en el compromiso, proliferación, diferenciación y extravasación de los leucocitos y sus progenitores en cada uno de estos compartimientos. Los mecanismos que producen la leucocitosis varían según el fondo común de leucocitos afectado y según cada factor en particular. Tenemos 5 (cinco) tipos principales de leucocitosis. Puede ser reflejo de un aumento de la población de neutrófilos (neutrofilia), linfocitos (linfocitosis), o monocitos (monocitosis). Rara vez, un aumento de eosinófilos y basófilos es tan grande como para ocasionar una leucocitosis. Es igualmente infrecuente que todas las líneas celulares estén aumentadas al mismo tiempo. Las infecciones y otros estados inflamatorios son capaces de causar no solo leucocitosis, sino también de afectar a los ganglios linfáticos. Hay muchas infecciones causantes de linfadenitis, algunos producen cuadros característicos, pero la gran mayoría son inespecíficos así que por eso lo denominamos Linfadenitis inespecífica la cual puede ser AGUDA o CRÓNICA.
 tipos principales de leucocitosis. Puede ser reflejo de un aumento de la población de neutrófilos (neutrofilia), linfocitos (linfocitosis), o monocitos (monocitosis). Rara vez, un aumento de eosinófilos y basófilos es tan grande como para ocasionar una leucocitosis. Es igualmente infrecuente que todas las líneas celulares estén aumentadas al mismo tiempo. Las infecciones y otros estados inflamatorios son capaces de causar no solo leucocitosis, sino también de afectar a los ganglios linfáticos. Hay muchas infecciones causantes de linfadenitis, algunos producen cuadros característicos, pero la gran mayoría son inespecíficos así que por eso lo denominamos Linfadenitis inespecífica la cual puede ser AGUDA o CRÓNICA.")
15
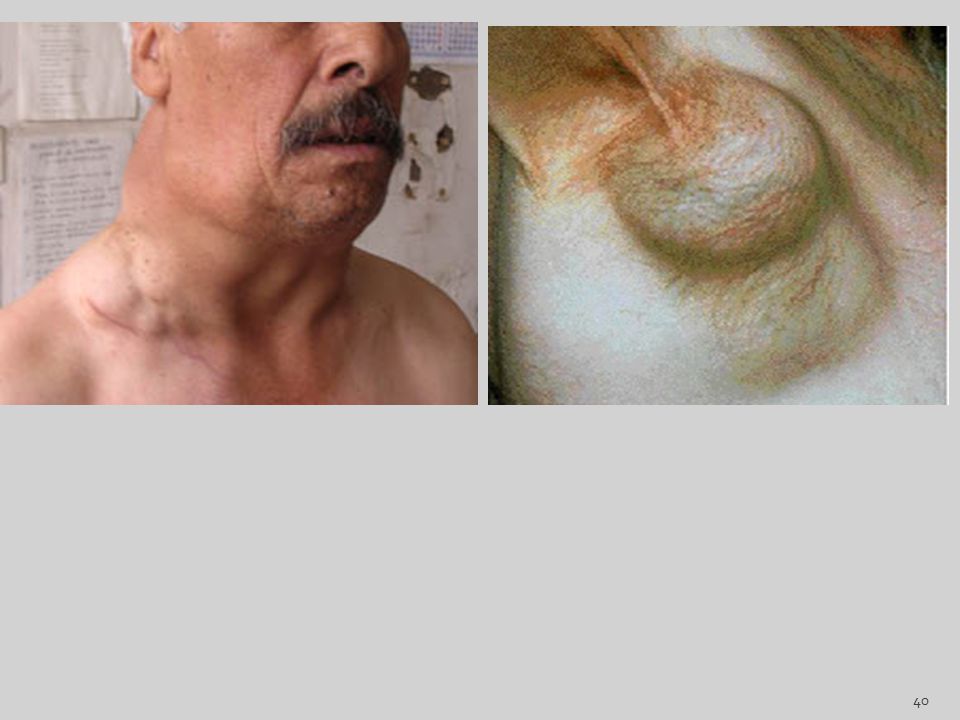
Linfadenitis Aguda La inflamación aguda de los ganglios se debe casi siempre al drenaje directo de los microorganismos y se observa en la región cervical, también en las regiones axilares o inguinales como consecuencia de infecciones de los miembros.

16
La inflamación se observa principalmente en la región cervical, acompañando las infecciones dentales o amigdalares. En los niños sobre todo, es frecuente que las infecciones virales y las bacteriemias produzcan adenopatías generalizadas. Los Ganglios están hinchados e ingurgitados y son de color gris rojizo. Clínicamente, los ganglios afectados aumentan de tamaño debido a la infiltración celular y al edema, se vuelve doloroso con la palpación debido a la distensión de la cápsula. La piel que lo cubre se enrojece con frecuencia y si la infección se extiende a la piel, aparecen fístulas especialmente cuando los ganglios linfáticos sufren necrosis con supuración.

17
Linfadenitis Crónica. Las reacciones crónicas de los ganglios linfáticos adoptan tres formas, según su etiología: Hiperplasia Folicular: se debe a un proceso inflamatorio que activa a las células B. Algunas causas especificas de hiperplasia folicular son la artritis reumatoide, la toxoplasmosis y los primeros estadios de la infección por VIH. Hiperplasia linfoide paracortical: Se caracteriza por que los cambios reactivos se producen en los lugares del ganglio ocupados por las células T. Hay además hipertrofia de las células endoteliales de los vasos y sinusoides. Estos cambios aparecen en las reacciones inmunitarias inducidas por fármacos, en las infecciones virales agudas, y especialmente en la Mononucleosis infecciosa. Hiperplasia Reticular: Se refiere a la distensión y resalte de los sinusoides linfáticos. Es una forma inespecífica de hiperplasia, pero puede ser especialmente acusada en los ganglios de drenaje de un cáncer, como el carcinoma de mama. Es característico de los ganglios linfáticos de las reacciones crónicas no sean dolorosos, porque la capsula del ganglio no soporta ningún aumento de presión. Es especialmente frecuente en los ganglios inguinales y axilares.

18
Leucopenia. Es la disminución anormal del numero de glóbulos blancos, por debajo de 5000/ mm³. Este trastorno puede afectar a cualquiera de los tipos específicos de los GB pero con mayor frecuencia afecta a los neutrófilos.

19
Neutropenia (agranulocitosis)
Es la disminución de los Neutrófilos. Se define como un recuento de Neutrófilos circulantes menor de 1500 células/uL. La reducción de los granulocitos puede deberse a una producción reducida o inefectiva de Neutrófilos o bien a una eliminación excesiva de Neutrófilos de la sangre.

20
Se observa granulopoyesis insuficiente o ineficaz en los siguientes casos:
Inhibición de las células madres mieloides. Inhibición de los precursores comprometidos de los granulocitos. Procesos patológicos caracterizados por granulopoyesis ineficaz. Procesos hereditarios raros en los que os defectos genéticos de determinados genes dan lugar a una diferenciación granulocítica alterada.

21
La destrucción o eliminación acelerada de los Neutrófilos se observa en:
Agresiones a los neutrófilos mediadas por mecanismo inmunitario. Secuestro esplénico, donde la destrucción excesiva es secundaria al aumento de tamaño del bazo, lo que puede acompañarse también de mayor destrucción de los hematíes y plaquetas. Aumento de su utilización en la periferia.

22
Neutropenia adquirida..
La granulopoyesis puede deteriorarse como resultado de una variedad de trastornos en la medula ósea (como estuvimos viendo en las diapositivas anteriores). El sobrecrecimiento de células neoplásicas también puede suprimir la función de los precursores de los neutrófilos. Debido a que la vida del neutrófilo dura alrededor de 1 día en la sangre periférica, la neutropenia aparece con rapidez cuando se deteriora la granulopoyesis. En estas condiciones la neutropenia suele acompañarse de trombocitopenia. En la anemia aplásica todas las células madres mieloides están afectadas, lo que produce anemia, trombocitopenia y agranulocitosis.
. El sobrecrecimiento de células neoplásicas también puede suprimir la función de los precursores de los neutrófilos. Debido a que la vida del neutrófilo dura alrededor de 1 día en la sangre periférica, la neutropenia aparece con rapidez cuando se deteriora la granulopoyesis. En estas condiciones la neutropenia suele acompañarse de trombocitopenia. En la anemia aplásica todas las células madres mieloides están afectadas, lo que produce anemia, trombocitopenia y agranulocitosis.")
23
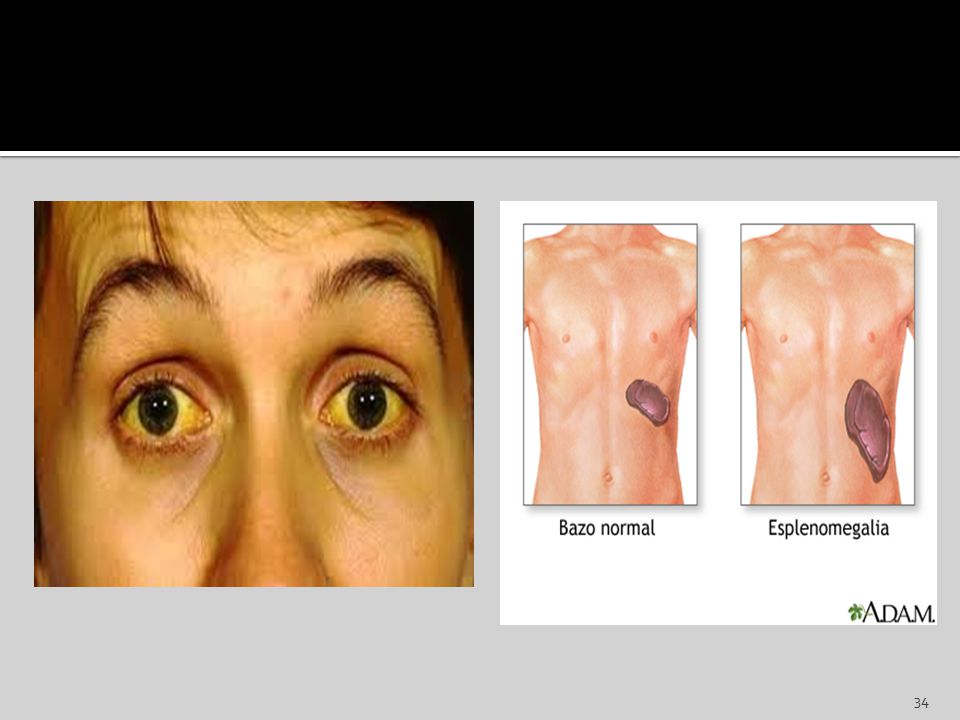
Los trastornos autoinmunitarios y las reacciones medicamentosas idiosincrásicas pueden causar mayor destrucción prematura de neutrófilos. En la esplenomegalia los neutrófilos pueden ser atrapados en el bazo junto con las otras células sanguíneas. En la enfermedad de Flety, por ejemplo, hay un aumento de la destrucción de los neutrófilos en el bazo.

24
Entre todas las asociaciones mencionadas, las neutropenias mas importantes son las debidas a fármacos. Algunos medicamentos como los agentes alquilantes y los antimetabolitos utilizados para combatir el cáncer, producen agranulocitosis de un modo previsible y relacionado con la dosis. Varios fármacos como el antibiótico cloranfenicol, los tranquilizantes fenotiazínicos, las sulfonamidas, el propiltiouracilo y la fenilbutazona pueden causar depresión idiosincrásica de la función de la médula ósea.

25
Otros fármacos como los derivados de la hidantoína y la primidona, pueden causar la destrucción intramedular de los granulocitos y en consecuencia dañar la producción de esas células.

26
Neutropenia Congénita.
La producción disminuida de granulocitos es una característica de un grupo de trastornos hematológicos hereditarios, entre los que figuran la neutropenia cíclica y el síndrome de Kostmann. La neutropenia cíclica o periódica es un trastorno autosómico dominante con expresión variable que comienza en la infancia, persiste durante décadas y se caracteriza por un cuadro de neutropenia periódica que se desarrolla cada 21 a 30 días y dura entre 5 a 6 días. La causa no ha sido determinada. El Síndrome de Kostmann es un trastorno autosómico recesivo, causa neutropenia intensa pero con preservación del linaje de células eritroides y megacariociticas. El recuento total de GB puede estar dentro de los límites normales, pero el neutrófilo es menor de 200/ul. Los niveles de monocitos y eosinófilos pueden ser elevados. En los neonatos de mujeres hipertensas puede existir una neutropenia transitoria, cuadro que suele durar de 1 a 60hs pero puede persistir durante 3 a 30 días.

27
Evolución clínica. Depende de la intensidad de la neutropenia y de la causa del trastorno. En las personas con neutropenia son comunes las infecciones, las infecciones que podrían pasar inadvertidas en personas con un recuento normal de neutrófilos podrían ser fatales en una persona con neutropenia. Los signos y síntomas iniciales son los de las infecciones bacterianas o micóticas e incluyen malestar Gral., escalofríos y fiebre, seguidos de debilidad extrema y fatiga. El sitio mas frecuente de infección grave es el tracto respiratorio. Son comunes las lesiones necrosantes ulcerativas de la boca, también pueden aparecer ulceraciones en la piel, vagina, y tracto gastrointestinal. Los antibióticos se utilizan cuando hay fiebre y para tratar las infecciones en situaciones en las que es posible controlar la destrucción de neutrófilos. Después de administrar un ciclo de quimioterapia mielodepresora, se puede utilizar el GM – CSF (Factor estimulador de colonias de granulocitos y macrofagos) con el fin de estimular la maduración y diferenciación del linaje de células granulocítica.
 con el fin de estimular la maduración y diferenciación del linaje de células granulocítica.")
28
Mononucleosis Infecciosa.
Es un trastorno que afecta al sistema linfomononuclear autolimitado causado por el virus de Epstein-Barr (EBV), miembro de la familia herpesvirus. Es un virus de distribución mundial, es mas frecuente en adolescentes y los adultos jóvenes pertenecientes a clases socioeconómicas altas en los países desarrollados. Este fenómeno probablemente se deba a que la enfermedad, que es relativamente asintomática durante la niñez, confiere inmunidad completa contra el virus.
, miembro de la familia herpesvirus. Es un virus de distribución mundial, es mas frecuente en adolescentes y los adultos jóvenes pertenecientes a clases socioeconómicas altas en los países desarrollados. Este fenómeno probablemente se deba a que la enfermedad, que es relativamente asintomática durante la niñez, confiere inmunidad completa contra el virus.")
29
Patogenia: Es transmitida sobre todo a través del contacto oral con saliva contaminada por EBV. El virus penetra en las células epiteliales nasofaríngeas, orofaríngeas y salivales, luego se disemina al tejido linfoide orofaríngeo subyacente y mas específicamente a los linfocitos B, que son los que tienen receptores para EBV. Las infecciones de las células B pueden adoptar una de las dos formas posibles: Pueden destruir las células infectadas o pueden incorporarse a su genoma.

30
En la mayor parte de los casos el virus se asocia con el genoma de la célula B. Las células que albergan el genoma del EBV proliferan en la circulación y producen anticuerpos heterófilos (que se utilizan para el diagnostico de la Mononucleosis infecciosa). La respuesta inmunitaria NORMAL es importante para controlar la proliferación de las células B infectadas por EBV. Las células más importantes para controlar la proliferación de las células B infectadas por EBV son los linfocitos T citotóxicos CD8 y los linfocitos NK. Aunque las células B infectadas y los viriones libres desaparecen de la sangre luego que el paciente se recupero, el virus permanece en algunas pocas células B transformadas en la región orofaringea y se elimina hacia la cavidad bucal, donde es vehiculizado a través de la saliva a otro hospedador.

32
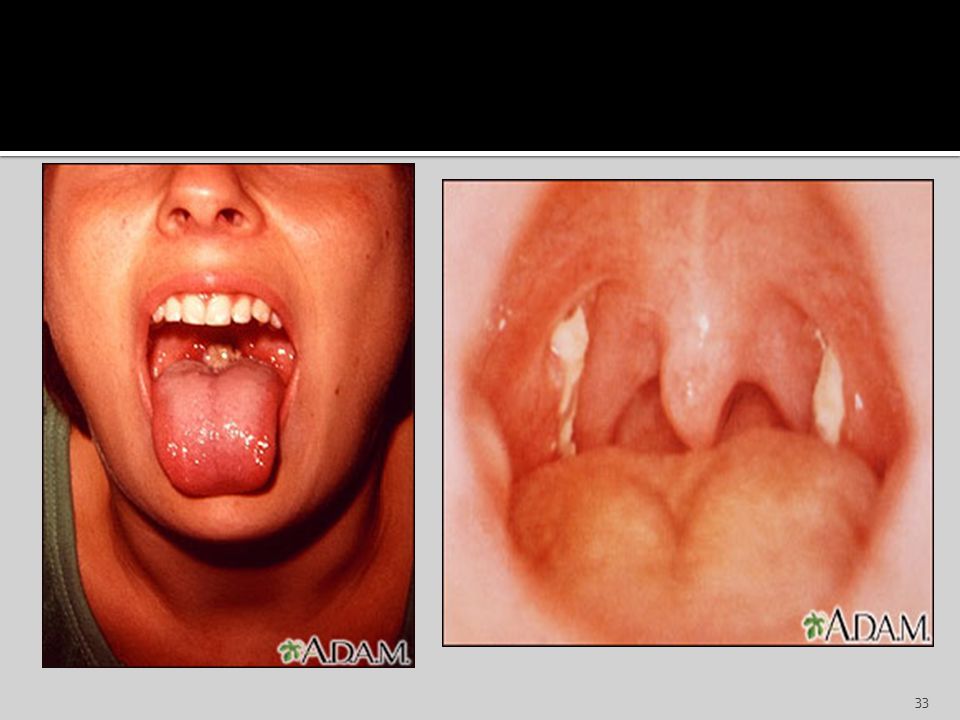
Evolución clínica: El periodo de incubación dura de 4 a 8 semanas
Evolución clínica: El periodo de incubación dura de 4 a 8 semanas. Luego sigue el periodo prodrómico que dura varios días y se caracteriza por malestar en general, anorexia y escalofríos. El periodo prodrómico precede al comienzo de la fiebre, faringitis y linfadenopatía. La inflamación de ganglios se da en todo el organismo, en particular en la región cervical, axilar e inguinal. Son característicos de esta enfermedad la Hepatitis y la Esplenomegalia. La hepatitis se caracteriza por: Hepatomegalia, nauseas, anorexia e ictericia. El bazo puede alcanzar un tamaño dos a tres veces mayor a lo normal y su rotura es una complicación poco frecuente. En el 10 a 15 % se desarrolla una erupción similar a la Rubeola, en menos del 1% se desarrollan complicaciones del SNC, estas complicaciones incluyen: parálisis de los nervios craneanos, encefalitis, meningitis, mielitis transversa y sx de Guillan-Barré.

35
Por lo general la sangre periférica muestra un aumento del número de leucocitos, con recuento de glóbulos blancos de entre y /ul. El 95% de los cuales son linfocitos. Si bien la leucocitosis es común, en algunas personas puede observarse leucopenia durante los 3 primeros días de la enfermedad. Los anticuerpos Heterófilos habitualmente aparecen a la segunda o la tercera semana y declinan después de finalizar la fase aguda. Sin embargo pueden ser detectables hasta 9 meses después del comienzo del trastorno. La mayoría de las personas con Mononucleosis infecciosa se recuperan sin secuelas. Es posible que persista cierto grado de debilidad y letargo durante 2 a 3 meses. El tratamiento: es sintomático y de sostén. Básicamente reposo en cama, analgésicos.

36
Linfomas Los linfomas son canceres de los linfocitos, los cuales residen en el sistema linfático y en los órganos formadores de sangre . Los linfomas pueden desarrollarse tanto a partir de los linfocitos B como de los linfocitos T. Los linfocitos T son importante para regular el sistema inmunológico y las infecciones víricas. Los linfocitos B producen anticuerpos. Los ganglios linfáticos diseminados por toda la red de vasos linfáticos, albergan un gran numero de linfocitos. Los linfocitos que se vuelven cancerosos (células de linfomas), pueden permanecer limitados a un solo ganglio linfático o pueden extenderse a la médula ósea, al bazo o prácticamente a cualquier otro órgano. Los dos tipos principales de linfomas son los linfomas de Hodgkin, más comúnmente conocidos como enfermedad de Hodgkin y linfoma no Hodgkin. El linfoma no Hodgkin es una enfermedad mucho más común que la enfermedad de Hodgkin. El linfoma de Burkitt y la Micosis fungoide son subtipos del linfima no Hodgkiniano.
, pueden permanecer limitados a un solo ganglio linfático o pueden extenderse a la médula ósea, al bazo o prácticamente a cualquier otro órgano. Los dos tipos principales de linfomas son los linfomas de Hodgkin, más comúnmente conocidos como enfermedad de Hodgkin y linfoma no Hodgkin. El linfoma no Hodgkin es una enfermedad mucho más común que la enfermedad de Hodgkin. El linfoma de Burkitt y la Micosis fungoide son subtipos del linfima no Hodgkiniano.")
37
Enfermedad de Hodgkin La enfermedad de Hodgkin es un tipo de linfoma caracterizado por la presencia de una clase particular de célula cancerosa llamada célula de Reed-Stenberg. La enfermedad es mas común en varones que en mujeres. Su máxima incidencia aparece en la juventud entre los 20 a 35 años y en mayores de 60 años. la causa de la enfermedad de Hodgking es desconocida. Hay una gran evidencia de que, en algunas personas la infección por el virus de Epstein-Barr es la causa de que los linfocitos B se vuelvan cancerosas y se transforman en células Reed-Stenberg. Aunque existen casos familiares, la mayoría de los casos con enfermedad de Hodgkin no tienen una base hereditaria.

38
Células de Reed-Stenberg.
Son multinucleadas o tienen un núcleo celular bilobulado, dando la apariencia de los ojos de búho, con nucléolos prominentes en forma de inclusiones.

39
Síntomas. Enf. De Hodgkin
Debilidad y dificultad respiratoria, como resultado de un número escaso de glóbulos rojos; infección y fiebre, como consecuencia de la disminución de la cifra de G.B.; y hemorragias producidas por una reducción del número de plaquetas; puede aparecer dolor óseo. Pérdida de la fuerza muscular, ronquera. Ictericia Inflamación de la cara, el cuello y las extremidades superiores (Síndrome de la Vena cava superior). Inflamación de las piernas y de los pies. Tos y dificultad respiratoria. Reducción de la capacidad para combatir las infecciones e incremento de la susceptibilidad a padecer infecciones producidas por hongos y virus.
. Inflamación de las piernas y de los pies. Tos y dificultad respiratoria. Reducción de la capacidad para combatir las infecciones e incremento de la susceptibilidad a padecer infecciones producidas por hongos y virus.")
41
Diagnostico Ante la sospecha clínica de una enfermedad de Hodgkin se deben solicitar las siguientes pruebas complementarias para la confirmación diagnóstica y estadificación: Hemograma Radiografía de tórax Biopsia de un ganglio linfático . Biopsia de médula ósea: Está indicada sobre todo en los estadios IB-IIB y III y IV. Determinación del VIH, debido a la infrecuente asociación con el sida.

42
Probabilidad de curacion
Estadios Los estadios de la enfermedad de Hodgkin son cuatro: I, II, III y IV, que se subclasifican en categoría A si no tienen síntomas y categoría B si presentan dichos síntomas. Cuando presentan extensión extralinfática se clasifican con la letra E. estadio GRADO DE DISEMINACION Probabilidad de curacion I II III IV Limitado a un solo ganglio linfatico. Afecta dos o mas ganglios en el mismo lado del diafragma, por encima o por debajo de éste (ej.: aumento de tamaño en algunos ganglios en el cuello y en la axila). Afecta a los ganglios linfaticos tanto por encima como por debajo del diafragma (por ej.: aumento de tamaño de algunos ganglios en el cuello y algunos en la ingle). Afecta también a otros órganos (tales como la médula ósea, los pulmones o el higado). Más del 95% 90% 80% Del 60 al 70%
. Afecta a los ganglios linfaticos tanto por encima como por debajo del diafragma (por ej.: aumento de tamaño de algunos ganglios en el cuello y algunos en la ingle). Afecta también a otros órganos (tales como la médula ósea, los pulmones o el higado). Más del 95% 90% 80% Del 60 al 70%")
43
Tratamiento y pronóstico
El manejo general de la enfermedad de Hodgkin consiste en: En estadios iniciales sin enfermedad masiva, se aplicará tratamiento combinado consistente en pocos ciclos de quimioterapia más radioterapia sobre regiones ganglionares afectadas. En pacientes con pronóstico intermedio o estadio II con enfermedad masiva mediastínica, se administrará tratamiento combinado de quimio y radioterapia. En estadios III y IV, se administrará tratamiento sistémico de quimioterapia con o sin radioterapia sobre regiones ganglionares afectas. Un posible efecto secundario del tratamiento es la pérdida de la capacidad reproductora. Sin embargo, aunque existe el riesgo de perder la capacidad de tener hijos (debido fundamentalmente a que la quimio y radioterapia pueden destruir los tejidos reproductores), existen en la actualidad diferentes medios de preservación de fertilidad
, existen en la actualidad diferentes medios de preservación de fertilidad.")
44
Linfoma No Hodgkin Los linfomas no Hodgkin constituyen un grupo diverso de cánceres que se desarrolla en los linfocitos B o T. Este grupo de cánceres corresponde realmente a un grupo de mas de 20 enfermedades diferentes, las cuales también difieren en su aparición al microscopio, en sus patrones celulares y en su curso clínico. La mayoría son de las células B. menos del 15% se desarrollan a partir de las células T. Es mas común que la enfermedad De Hodgkin y la incidencia se esta incrementando, en especial, entre las personas mayores y entre las que presentan un sistema inmunológico deficiente. Aunque se desconoce la causa del linfomano Hodgkin, hay una fuerte evidencia que respalda el papel de los virus en algunos de los tipos menos comunes de este linfoma.

45
Síntomas Dificultad para respirar, inflamación en la cara.
Pérdida de apetito, estreñimiento severo, dolor abdominal o distención. Inflamación intensiva de las piernas. Pérdida de peso, diarrea, mal absorción. Acumulación de liquidos alrededor de los pulmones (derrame pleural). Áreas de piel engrosada, oscura y pruriginosa. Fiebre y sudoración nocturna. Anemia Susceptibilidad a padecer infecciones bacterianas graves.
. Áreas de piel engrosada, oscura y pruriginosa. Fiebre y sudoración nocturna. Anemia. Susceptibilidad a padecer infecciones bacterianas graves.")
46
Diagnostico y clasificación
Se debe realizar la biopsia de un ganglio linfático agrandado para diagnosticar y diferenciar el linfoma no Hodgkin de la enfermedad de Hodgkin o de otros problemas que cursan con agrandamiento de los ganglios linfaticos. Aunque más de 20 enfermedades diferentes pueden denominarse linfomas no Hodgkin se les clasifica de la siguiente manera: Indolentes Agresivos Muy agresivos ESTADIOS normalmente en el momento del diagnostico la enfermedad ya se encuentra diseminada. Si se diagnostica tempranamente se puede comprobar que tiene estadios semejantes a la enfermedad de Hodgkin.

47
Tratamiento y pronóstico
Estadios I y II. En los linfomas de bajo grado, la radioterapia regional permite el control a largo plazo. linfomas de grado intermedio quimioterapia y radioterapia localizada; con esto se curan el 70 al 90% de las personas en este estadio. Estadios III y IV. Las personas que cursan por este estadio no siempre requieren tratamiento, pero si son controladas de cerca para poder evidenciar complicaciones. Si la enfermedad comienza a avanzar más rápidamente, hay muchas opciones de tratamiento. El tratamiento Puede incluir quimioterapia con una sola droga o esquemas combinados en los que se emplean varias drogas. El tratamiento logra una remisión, pero su promedio de duración oscila entre 2 a 4 años. Después de una recaída las remisiones tienden a volverse mas cortas. En los pacientes con linfomas muy agresivos se administra quimioterapias que componen la combinación de varios fármacos. Ej.: CHOP (ciclofosfamida, hidroxi doxorubicina, vincristina y prednisona). El esquema CHOP con rituximab pueden ser mejores. Trasplante de células madres.
. El esquema CHOP con rituximab pueden ser mejores. Trasplante de células madres.")
48
Leucemia Son canceres de los glóbulos blancos, o de las células que maduran a glóbulos blancos. Los glóbulos blancos se originan a partir de las células madre en la médula ósea. A veces se producen errores en su maduración, y algunos fragmentos de los cromosomas se reorganizan. Los cromosomas anormales resultantes afectan al control normal de la división celular y hacen que las células se multipliquen sin control y se conviertan en cancerosas. Estas células malignas también pueden ir invadiendo otros órganos, como el hígado, el bazo, los ganglios linfáticos, los testículos y el cerebro. Las causas aun se desconocen. La exposición a la radiación o a ciertos tipos de quimioterapia incrementa el riesgo a desarrollar algunas formas de leucemia. Ciertos trastornos hereditarios como el síndrome de Down y el síndrome de Fanconi, también aumentan el riesgo. Las leucemias se clasifican en 4 grupos según la rapidez con que evolucionan: leucemia linfocítica aguda y leucemia mielocítica aguda. Leucemia linfocítica crónica y leucemia mielocíticaa crónica.

49
Leucemia linfocítica aguda
La LLA es una enfermedad que puede poner en peligro la vida y en la cual las células que normalmente se convierten en linfocitos se transforman en cancerosas y reemplazan a las células normales que se encuentran en la médula ósea. Esta aparece en cualquier edad, aunque sea más frecuente en menores de 15 años. El la LLA las células leucémicas inmaduras se acumulan en la médula ósea, destruyendo y reemplazando a las que producen células sanguíneas normales. Las células leucémicas también son transportadas por el torrente sanguíneo hacia los órganos donde pueden continuar su crecimiento y división. Pueden provocar irritación de la membrana que recubre el cerebro y la médula espinal causando inflamación (meningitis), anemia e insuficiencia hepática y renal, y causar daños en otros órganos.
, anemia e insuficiencia hepática y renal, y causar daños en otros órganos.")
50
Síntomas y diagnostico
Fiebre y sudoración excesiva. Debilidad, fatiga y palidez, indicios de anemia. Sangrado nasal o de encías. Dolor de cabeza, vomito e irritabilidad. Dolores óseos y de las articulaciones. Cuando las células leucémicas agrandan el hígado y el bazo se puede presentar sensación de plenitud abdominal, y en algunos casos, dolor.

51
Los análisis de sangre con un recuento complementario de células sanguíneas pueden proporcionar la primera prueba de la presencia de la LLA. El número total de glóbulos blancos puede ser bajo normal o elevado pero la cantidad de glóbulos rojos y plaquetas esta siempre disminuida. Además en las muestras de sangre observadas al microscopio pueden observarse G.B. inmaduros (blastos). Para lograr diferenciar una LLA de otros tipos de leucemias debe realizarse una biopsia de la médula ósea. Un examen físico puede revelar lo siguiente: Hematomas Hepatomegalia, esplenomegalia e inflamación de los ganglios linfáticos Signos de sangrado (petequias, púrpura)
")
52
Tratamiento El primer objetivo del tratamiento es lograr que los conteos sanguíneos vuelvan a la normalidad. Si esto ocurre y la médula ósea se ve saludable bajo el microscopio, se dice que el cáncer está en remisión. Después de la remisión, se necesitará más tratamiento para curarse. Este tratamiento puede incluir más quimioterapia o un trasplante de células madre de un donante. La quimioterapia es el primer tratamiento empleado para intentar que la enfermedad entre en remisión. La primera vez se suministre la quimioterapia, el paciente necesita permanecer en el hospital durante varias semanas. Posteriormente, puede recibir la quimioterapia en forma ambulatoria. Si tiene un conteo de linfocitos bajo, es posible que sea necesario dejarlo solo en un cuarto del hospital para que no contraiga una infección de otras personas. La leucemia linfocítica aguda se puede propagar al cerebro y la médula espinal. Muchos fármacos quimioterapéuticos administrados por vía intravenosa no pueden llegar hasta estas áreas; por lo tanto, debe suministrarse: Quimioterapia directamente dentro del espacio alrededor del cerebro y la columna vertebral. Radioterapia al cerebro.

53
Leucemia mielocítica aguda
La leucemia mielocítica aguda es una enfermedad potencialmente mortal en la cual las células que se convierten en neutrófilos, basófilos, eosinófilos y monocitos se transforman en cancerosas y reemplazan rápidamente a las células normales de la médula ósea. La LMA es el tipo mas común de leucemia en la población adulta. Las células leucémicas pueden formar pequeñas masas (cloromas) dentro o debajo de la piel, en las encías y en los ojos. La leucemia promielocítica aguda es un subtipo de LMA. En esta variante, las anomalías cromosómicas de los promielocítos (que son las células en una etapa temprana del proceso de maduración que las transformará en neutrófilos) impidiendo la unión y la actividad de la vitamina A. sin ella se interrumpe la maduración normal de las células y se acumulan en promielocítos anormales.
 dentro o debajo de la piel, en las encías y en los ojos. La leucemia promielocítica aguda es un subtipo de LMA. En esta variante, las anomalías cromosómicas de los promielocítos (que son las células en una etapa temprana del proceso de maduración que las transformará en neutrófilos) impidiendo la unión y la actividad de la vitamina A. sin ella se interrumpe la maduración normal de las células y se acumulan en promielocítos anormales.")
54
Síntomas y diagnósticos
Los primeros síntomas de la LMA son muy similares a los de la LLA. Aunque la meningitis es menos frecuente. El diagnostico de la LMA también es similar al de la LLA. Para hacer la diferenciación de los diversos tipos de leucemia casi siempre es necesario someterse a una biopsia de médula ósea.

55
Pronóstico y tratamiento
Sin tratamiento, la mayoría de las personas con LMA mueren a las pocas semanas o meses del diagnostico. Con tratamiento es posible prolongar ese tiempo en hasta cinco años, sin sufrir recaidas. El tratamiento de la LMA se basa principalmente en la quimioterapia y está dividido en dos fases, terapia de inducción y terapia de posremisión (o consolidación). El objetivo de la terapia de inducción es llevar a cabo una reducción del número de células leucémicas hasta niveles indetectables. El objetivo de la terapia de consolidación es la completa eliminación de cualquier resto de la enfermedad y lograr la curación completa del paciente
. El objetivo de la terapia de inducción es llevar a cabo una reducción del número de células leucémicas hasta niveles indetectables. El objetivo de la terapia de consolidación es la completa eliminación de cualquier resto de la enfermedad y lograr la curación completa del paciente.")
56
Leucemia linfocítica crónica
La LLC es una enfermedad en la cual los linfocitos maduros se convierten en cancerosos y gradualmente reemplazan a las células normales de los ganglios linfáticos. Más de tres cuartas partes de la personas que padecen LLC son mayores de 60 años, y la enfermedad no se presenta en niños. Es de mayor incidencia en varones. Es común en América del Norte y Europa, lo cual indica que la herencia desempeña una función importante en su aparición.

57
se caracteriza por la proliferación de linfocitos morfológicamente maduros pero inmunológicamente inmaduros y se manifiesta por su acumulación progresiva en la sangre, médula ósea y tejido linfático. Aunque es incurable, su evolución, en la mayoría de los casos es indolente durante años, sin embargo, en algunos individuos la supervivencia es de sólo unos meses. La hemorragia y la infección son una causa principal de muerte en estos pacientes. Los factores pronósticos incluyen el subgrupo citogenético, estado mutacional de inmunoglobulina, ZAP-70 y CD38. Ya que estos pacientes tienen un aumento en el riesgo de presentar otras neoplasias, aún antes del tratamiento y los avances genéticos e Inmunohistoquímicos.

58
Síntomas y diagnósticos
Los síntomas que pueden presentarse abarcan: Hematomas anormales (ocurre en las últimas etapas de la enfermedad) Inflamación de los ganglios linfáticos, el hígado o el bazo Sudoración excesiva, sudores fríos Fatiga Fiebre Infecciones que siguen reapareciendo (recurrentes) Inapetencia o sentir llenura con demasiada rapidez (saciedad temprana) Pérdida de peso involuntaria Por lo regular tienen un conteo de glóbulos blancos más alto de lo normal. La biopsia de médula ósea no suele ser muy necesario, porque pueden llevarse a cabo exámenes especializados de las células para catalogar los linfocitos.
 Inflamación de los ganglios linfáticos, el hígado o el bazo. Sudoración excesiva, sudores fríos. Fatiga. Fiebre. Infecciones que siguen reapareciendo (recurrentes) Inapetencia o sentir llenura con demasiada rapidez (saciedad temprana) Pérdida de peso involuntaria. Por lo regular tienen un conteo de glóbulos blancos más alto de lo normal. La biopsia de médula ósea no suele ser muy necesario, porque pueden llevarse a cabo exámenes especializados de las células para catalogar los linfocitos.")
59
Tratamiento Dado que LLC evoluciona lentamente, muchas personas no necesitan tratamiento durante años, hasta que el número de linfocitos comienza a aumentar y los ganglios linfáticos se empiezan a agrandar o el número de G.R. o plaquetas disminuye. Los fármacos utilizados en la leucemia ayudan a aliviar los síntomas y a eliminar el crecimiento de los ganglios linfáticos y el bazo, pero no curan la enfermedad. A la larga la LLC se vuelve se vuelve resistente a los fármacos utilizados inicialmente y algunas veces es necesario probar otros tratamientos. La anemia secundaria debe tratarse con transfusión de sangre y ocasionalmente con inyecciones de eritropoyetina. Los recuentos bajos de plaquetas se tratan con transfusiones de este tipo de células sanguíneas y las infecciones con antibióticos. La radioterapia se utiliza para la reducción del agrandamiento de los ganglios y demás órganos.

60
Leucemia mielocítica crónica
Es un síndrome mieloproliferativo crónico de naturaleza clonal, originada en la célula madre, que resulta en un excesivo número de células mieloides en todos los estadios de maduración. Fue la primera enfermedad maligna en que se demostró una anomalía genética adquirida y es en la actualidad el modelo molecular de leucemia mejor estudiado. Aparece una traslocación genética de tipo t(9;22)1 que produce un reordenamiento de los genes BCR/ABL, produciendo el denominado cromosoma Filadelfia2 descubierta en 1960 por Newell y Hungerford.3 La proteína que resulta es una tirosin quinasa cuya alteración transforma el ATP en ADP, fosforilando un sustrato que altera la médula ósea y su funcionamiento. Puede afectar a personas de cualquier edad y sexo, es más frecuente en adultas con edades comprendidas de entre los 40 y 60 años.
1 que produce un reordenamiento de los genes BCR/ABL, produciendo el denominado cromosoma Filadelfia2 descubierta en 1960 por Newell y Hungerford.3 La proteína que resulta es una tirosin quinasa cuya alteración transforma el ATP en ADP, fosforilando un sustrato que altera la médula ósea y su funcionamiento. Puede afectar a personas de cualquier edad y sexo, es más frecuente en adultas con edades comprendidas de entre los 40 y 60 años.")
61
Síntomas y diagnóstico
Fase crónica o mielocitaria Dura unos 4 a 5 años, aunque puede precederse de una fase previa asintomática, caracterizada sólo por la alteración genética. Puede ser asintomática y detectarse en pruebas analíticas rutinarias, o presentar los siguientes síntomas: - Síntomas de hipoxia tisular Síntomas derivados de la esplenomegalia: pesadez postprandrial, la saciedad precoz o fenómenos compresivos abdominales. Fase acelerada Dura unos 6 u 8 meses. No se conocen bien los factores que promueven la transición a las siguientes fases de la enfermedad, pero los estudios citogenéticos y moleculares muestran nuevas alteraciones: la aparición de un segundo cromosoma Filadelfia, de una trisomía del cromosoma 8 o de una deleción p17-.

62
El enfermo presenta fiebre, aumento de la anemia y sus consecuencias, dolores óseos...
En las pruebas analíticas aparece aumento de los basófilos (por aumento de blastos), hipereosinofilia, anemia y trombocitopenia. Como consecuencia, aparecen infecciones, trombosis y/o hemorragias. Fase de transformación a leucemia aguda (crisis blástica) Aparecen más de un >20% de blastos en médula ósea. Por alteración genética de la célula madre en estadios madurativos más precoces, la leucemia mieloide crónica da crisis clínicas similares a la leucemia aguda. El 80% de los casos evolucionan a leucemia mieloblástica aguda (LMA), y el 20% a leucemia linfoblástica aguda (LLA), con mejor pronóstico. La clínica es de curso tormentoso, con anemia severa, infecciones de repetición, hemorragias y trombos, alteraciones multiorgánicas por infiltración linfocítica, signos de leucostasia. La clínica es indistinguible de la de la leucemia aguda, y hay que hacer el diagnóstico diferencial por técnicas de biología molecular.
, hipereosinofilia, anemia y trombocitopenia. Como consecuencia, aparecen infecciones, trombosis y/o hemorragias. Fase de transformación a leucemia aguda (crisis blástica) Aparecen más de un >20% de blastos en médula ósea. Por alteración genética de la célula madre en estadios madurativos más precoces, la leucemia mieloide crónica da crisis clínicas similares a la leucemia aguda. El 80% de los casos evolucionan a leucemia mieloblástica aguda (LMA), y el 20% a leucemia linfoblástica aguda (LLA), con mejor pronóstico. La clínica es de curso tormentoso, con anemia severa, infecciones de repetición, hemorragias y trombos, alteraciones multiorgánicas por infiltración linfocítica, signos de leucostasia. La clínica es indistinguible de la de la leucemia aguda, y hay que hacer el diagnóstico diferencial por técnicas de biología molecular.")
63
Tratamiento Actualmente el tratamiento preferido para la LMC es el mesilato de imatinib, que inhibe la enzima tirosín-kinasa y ha permitido pasar de una esperanza de vida de cuatro años, a una cronificación de la enfermedad en el 90% de los casos. El imatinib, producido por Novartis con el nombre de Glivec o Gleevec, ha sido objeto de batallas judiciales y estudiado como ejemplo del alto precio de los medicamentos contra el cáncer. El trasplante de médula ósea (alogénico) es curativo, pero sólo se emplea en la fase acelerada.
 es curativo, pero sólo se emplea en la fase acelerada.")
64
Discrasia de los plasmocitos Expansión de un solo plasmocito productores de Ig y un aumento resultante de los niveles séricos de una sola Ig monoclonal o de sus fracciones. Las discrasias de los plasmocitos incluyen: Mieloma Múltiple. Mieloma Solitario. Linfoma Linfoplasmocitico. Amiloidosis primaria o inmunocítica. Gammapatía monoclonal de importancia indeterminada (GMII)
")
65
Mieloma Múltiple. Es sin duda la más frecuente de las discrasias malignas de los plasmocitos. Es más frecuente en personas mayores de 60 años y la edad promedio de los pacientes con mieloma múltiple es de 71 años. La aparición de la enfermedad es más frecuente en hombres que en las mujeres. La causa de Mieloma múltiple se desconoce. Los factores de riesgo incluyen la estimulación inmunitaria crónica, los trastornos autoinmunes, la exposición a radiación ionizante y exposición ocupacional a plaguicidas o herbicidas.

66
Patogenia: Se caracteriza por la proliferación de plasmocitos malignos en la médula ósea y lesiones osteolíticas en todo el sistema esquelético. Una de las características del mieloma es la producción NO regulada de un Ac monoclonal denominado proteína M debido a que se detecta como una espiga M en la electroforesis de la proteína. Las citocinas son importantes en la patogenia del trastorno . Son factores de crecimiento para la enfermedad: IL-6; IL-1; GM-CSF; INF-ALFA y la IL-10.

67
Manifestaciones: Los principales sitios afectados en los mielomas son los huesos y la médula ósea. Además de la proliferación anormal de plasmocitos medulares, hay proliferación de Osteoclastos, que predispone al paciente a fracturas patológicas e hipercalcemia. Las paraproteínas segregadas por los plasmocitos pueden causar hiperviscosidad de los líquidos corporales y pueden degradarse en amiloide, lo que causa insuficiencia cardíaca y neuropatía. Aunque el mieloma múltiple se caracteriza por la producción excesiva de inmunoglobulina monoclonal, los niveles de inmunoglobulinas normales suelen estar deprimidos. Los plasmocitos malignos también pueden formar plasmocitomas en el hueso y los tejidos blandos, el sitio mas común es el tracto gastrointestinal. Los plasmocitomas en el tejido óseo se asocian con destrucción del hueso y dolor localizado. Pueden observarse lesiones osteolíticas y fracturas por compresión en el esqueleto axial, y en la parte proximal de los huesos largos. El dolor óseo es uno de los primeros síntomas que se observan en las tres cuartas partes de todos los pacientes con mieloma múltiple, también Anemias e infecciones recurrentes, perdida de peso, debilidad, insuficiencia renal que se produce en el 50% de los casos

68
Diagnostico: Se basa en las manifestaciones clínicas, los análisis de sangre y el examen de la médula ósea. Las radiografías óseas, estudios del esqueleto y resonancia magnética son importantes para establecer la presencia de lesiones óseas.

69
Tratamiento: Melfalan y la prednisona, el agregado de antraciclinas, fármacos alquilantes alternativos e interferon solo permite lograr mejoras mínimas. La radioterapia solo sirve como tto de apoyo o alivio del dolor provocado por las lesiones osteolíticas y las fracturas por compresión. Tratamiento con Bisfosfonatos, también con eritropoyetina. Actualmente se considera que la quimioterapia en altas dosis es el tratamiento de elección para los pacientes menores de 70 años recién diagnosticados.

70
Gracias por su atención

Presentaciones similares


















>")
