Descargar la presentación
La descarga está en progreso. Por favor, espere

1
DETERIORO COGNITIVO SUBAGUDO
Dra. Diana Stancu ,MIR 1 MFyC Coordinador Dr. J.M.Moltó Servicio Neurología,Hospital Virgen de Los Lirios , Alcoi

2
Caso clínico: AP: Paciente de 55 años de edad con : HTA,No DM ,No DLP
IQ:amigdalectomizada Episodios vertiginosos Sdr. Ansioso-depresivo diagnosticado en diciembre de 2008 en tto con prozac En tto habitual con:parapres plus,coropres,nutrof 20,venoruton,orfidal,denubil Resto sin interes

3
Acude 2x a urgencias por: visión borrosa e inestabilidad de la marcha, temblor EESS(1/02/09) y agresividad y amenaza de muerte a su marido(19/02/09) 1/02/09 en urgencias exploración aumento de la base de sustentación, bradipsiquia; analitica:glucosa 133,ECG normal,resto normal; TAC craneal: discreta atrofia cerebral de predominio frontal 1/02/09-4/02/09: ingreso 4 dias en neurología

4
Valoración en Neurología
explor neur: habla escándida ,diplopia,aumento de la base de sustentación de la marcha,temblor telecinético en MSD,resto sin alteraciones RMN craneal: atrofia central simétrica desproporcionada a la de otras áreas analítica: colesterol 250,LDL167,glucosa basal 128,marcadores tumorales negativos dx al alta: probable síndrome cerebeloso otros dx: posible degeneración cortico basal/sdr ansioso depresivo de larga evolución

5
19/02/09: MC: agresividad Presenta: alteraciones de la visión, inestabilidad de la marcha, temblor EESS, mioclónias múltiples desencadenadas por estímulos auditivos Psiquiatría: lenguaje incoherente, afectividad inadecuada, llanto reactivo a discurso delirante de perjuicio, episodios de heteroagresividad física Según familia: rápido y grave deterioro, desorganización patrón sueño vigilia, conductas disruptivas, deshinhibición psicomotriz

6
Diagnóstico diferencial demencia subaguda:
Tumores primarios o metastásicos (RM) Complejo demencia-SIDA. (RM:imágenes hiperintensas periventriculares y atrofia cortico-subcortical) Hidrocefalia normotensiva. (RM:ventriculomegalia en sistema ventricular supra e infratentorial+clínica :apraxia marcha,incontinencía esfincteres,deterioro cognitivo) Encefalitis límbica. (RM:lóbulos temporales e inflamación LCR + clínica:días o semanas pérdida de memoria,convulsiones,confusión y alteraciones psiquiátricas+AC anti canales de K) Demencia fronto temporal. Degeneración córtico-basal. (Evolución rápida y clínica psiquiátrica). Enfermedad de Creutzfeldt Jakob (otras enf priónicas)
 Complejo demencia-SIDA. (RM:imágenes hiperintensas periventriculares y atrofia cortico-subcortical) Hidrocefalia normotensiva. (RM:ventriculomegalia en sistema ventricular supra e infratentorial+clínica :apraxia marcha,incontinencía esfincteres,deterioro cognitivo) Encefalitis límbica. (RM:lóbulos temporales e inflamación LCR + clínica:días o semanas pérdida de memoria,convulsiones,confusión y alteraciones psiquiátricas+AC anti canales de K) Demencia fronto temporal. Degeneración córtico-basal. (Evolución rápida y clínica psiquiátrica). Enfermedad de Creutzfeldt Jakob (otras enf priónicas)")
7
20/02/09-28/05/09 ingreso neurología
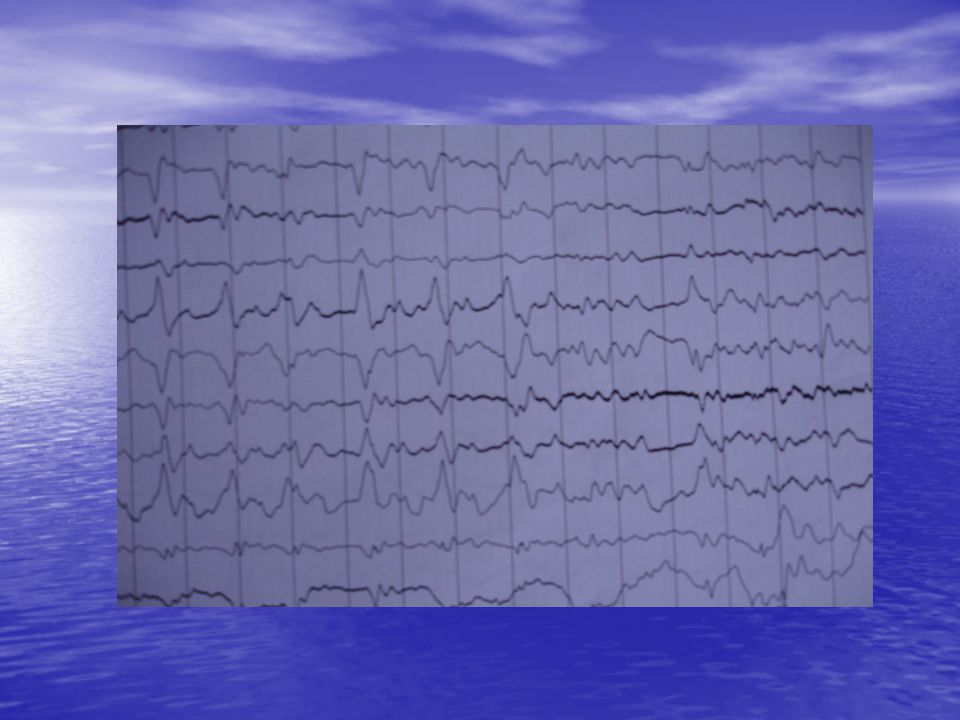
--durante ingreso: importante deterioro cognitivo, lenguaje desorganizado, conductas inapropiadas etc. --tto: neurolépticos sedativos --pruebas: Analitica sin hallazgos EEG: ondas muy lentas polimórficas a 1-3 c/s bifronto-temporales,con tendencia a generalización; signos de severa afectación encefálica difusa de predominio bifronto-temporal ** Trazado compatible con enf Creutzfeldt Jakob

9
RMN craneal: RMN : atrofia cortico subcortical en grado superior al presente en estudio previo

10
Estudio genético: mutación E200K: encefalopatía espongiforme transmisible de origen genético
Proteína en LCR positiva: ECJ esporádica 28/05/09: exitus por infección respiratoria (causa más frecuente de muerte) La familia rechaza estudio necrópsico
 La familia rechaza estudio necrópsico.")
11
Enfermedad priónica: Incidencia:1/millón de habitantes y año
Tipos: esporádica, yatrogénica, hereditaria y nueva variante Neurodegenerativa Largo período de incubación Rápidamente progresiva Enfermedad letal

12
Insomnio familiar fatal
espongiformes=aspecto característico de los cerebros infectados; se asemejan a esponjas bajo un microscopio HUMANAS: Kuru Insomnio familiar fatal La enfermedad de Gerstmann Straussler Scheinker La enfermedad de Creutzfeldt –Jakob Variante clásica Nueva variante

13
ANIMALES: scrapie encefalopatía espongiforme bovina (BSE) encefalopatía transmisible del visón enfermedad del desgaste crónico encefalopatía de ungulados exóticos encefalopatía espongiforme felina

14
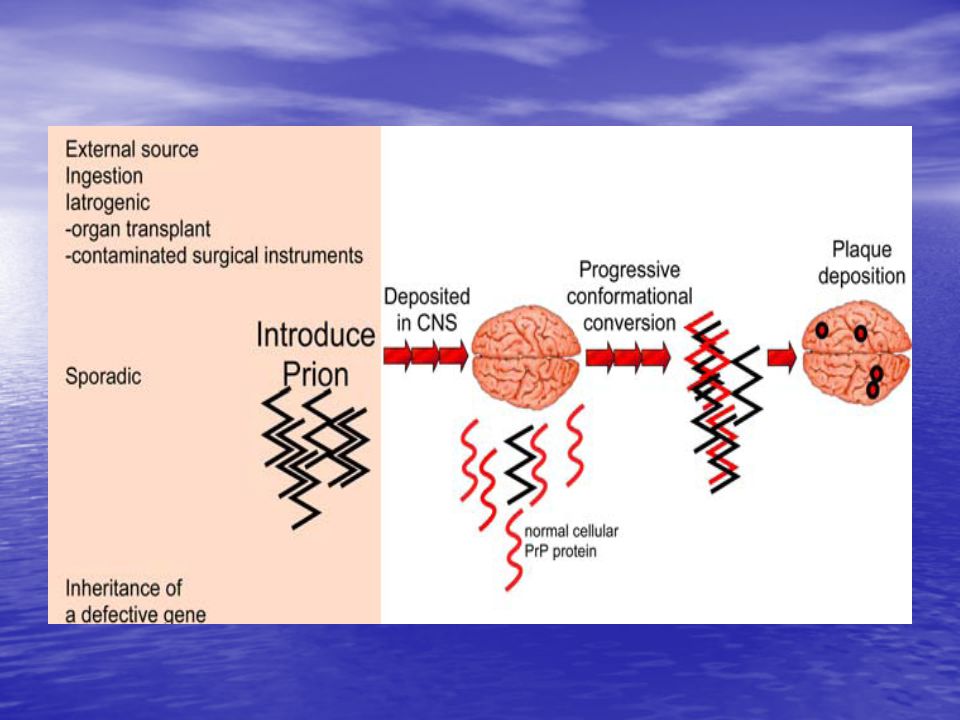
Son agentes transmisibles no convencionales carentes de ácidos nucleicos
El único componente conocido es una proteína endógena incorrectamente plegada (PrPSc) Presentan una gran resistencia a la mayoría de los procedimientos de desinfección tradicionales. No producen una respuesta inmune e inflamatoria relevante. En tejidos accesibles (sangre, orina) se encuentran en muy baja concentración Su distribución en distintos tejidos es irregular, expresándose principalmente en el SNC y tejido linforreticular Las enfermedades causadas por estos agentes tienen largos periodos de incubación
 Presentan una gran resistencia a la mayoría de los procedimientos de desinfección tradicionales. No producen una respuesta inmune e inflamatoria relevante. En tejidos accesibles (sangre, orina) se encuentran en muy baja concentración. Su distribución en distintos tejidos es irregular, expresándose principalmente en el SNC y tejido linforreticular. Las enfermedades causadas por estos agentes tienen largos periodos de incubación.")
15
Proteína priónica Hay 2 tipos: normal y anormal
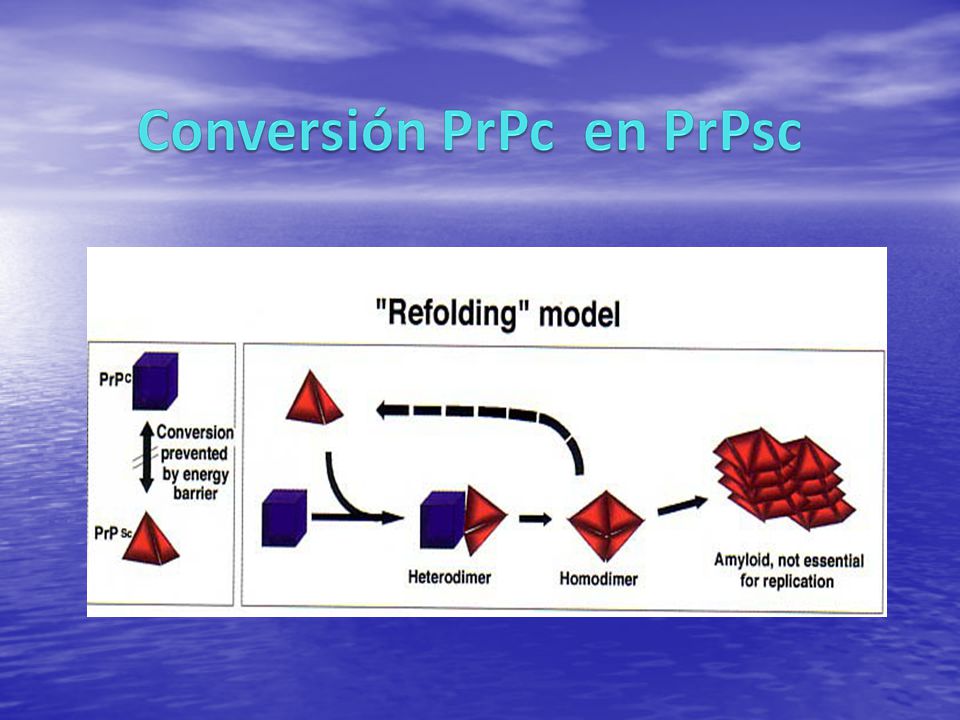
1.Normal(PrPc,codificada por un gen localizado en brazo corto del cromosoma 20): existe en el cerebro humano, otros mamíferos y pájaros su función es desconocida. 2.Anormal(PrP Sc): las proteínas anormales de los priones se unen y forman fibras o acumulaciones llamadas placas, que se pueden identificar en el cerebro años antes de que empiecen los síntomas. se puede formar por: conversión espontánea mutación genética conversión de la PrPc
: existe en el cerebro humano, otros mamíferos y pájaros. su función es desconocida. 2.Anormal(PrP Sc): las proteínas anormales de los priones se unen y forman fibras o acumulaciones llamadas placas, que se pueden identificar en el cerebro años antes de que empiecen los síntomas. se puede formar por: conversión espontánea. mutación genética. conversión de la PrPc.")
16
multimerisation state
PrPC PrPSc solubility soluble non soluble structure predominantly alpha-helical predominantly beta-sheeted multimerisation state monomeric multimeric (aggregates) infectivity non infectious infectious
 infectivity. non infectious. infectious.")
19
ECJ esporádica: 85% ECJ hereditaria: 5-10% ECJ adquirida:<1% ECJ nueva variante:<1%

20
Descrita por primera vez en 1920
Edad media de inicio : 68años Progresión rápida de los síntomas: -demencia -ataxia cerebelosa, falta de coordinación -mutismo acinético -movimientos bruscos,mioclonias Duración de la enfermedad: 4-5meses No hay factores de riesgo para la enfermedad conocidos

21
Historia familiar de la enfermedad o pruebas positivas de mutación genetica
Edad media de inicio: 52 años Síntomas similares a la forma esporádica Duración de la enfermedad: 4-5meses

22
Tiempo muy largo de incubación: aprox 30 años
No depende de la edad Tiempo muy largo de incubación: aprox 30 años Se transmite por exposición al tejido cerebral o del sistema nervioso, mediante procedimientos neuroquirúrgicos (transplante de córnea, equipamientos neuroquirúrgicos (lote de duramadre para plastias), hormona de crecimiento extraída de glándulas pituitarias de cadáveres etc). Accidental transmission of Creutzfeldt-Jakob disease by dural cadaveric grafts. Martínez-Lage JF, Poza M, Sola J, Tortosa JG, Brown P, Cervenáková L, Esteban JA, Mendoza A. J Neurol Neurosurg Psychiatry Sep;57(9): ¿CÓMO? Los priones sobreviven a los procedimientos de desinfección
, hormona de crecimiento extraída de glándulas pituitarias de cadáveres etc). Accidental transmission of Creutzfeldt-Jakob disease by dural cadaveric grafts. Martínez-Lage JF, Poza M, Sola J, Tortosa JG, Brown P, Cervenáková L, Esteban JA, Mendoza A. J Neurol Neurosurg Psychiatry Sep;57(9): ¿CÓMO Los priones sobreviven a los procedimientos de desinfección.")
23
Edad media de início: 27 años
En 1995 dos jóvenes en Reino Unido En 1996 el número aumenta a 10 casos Comienza principalmente con síntomas psiquiátricos y alteraciones de comportamiento precoces. Dominan los signos cerebelosos en la clínica neurológica. Evolución más prolongada: meses Transmisión: consumo de carne de vaca contaminada

24
Afectación de córtex cerebral y cerebeloso
Degeneración espongiforme Atrofia y pérdida de neuronas Gliosis astrocítica Ocasionalmente placas compactas de amiloide con acúmulos de PrP Característicamente no hay reacción inflamatoria

25
Cortex normal Cortex ECJ

26
Fases iniciales:-confusión,alucinaciones y agitación
-fallos en la memoria -depresión -cambios de comportamiento -falta de coordinación(ataxia cerebelosa) -alteraciones visuales(de la percepción o agnosias visuales) -astenia,anorexia,pérdida de peso , alteraciones de sueño o pérdida de libido.
 -alteraciones visuales(de la percepción o agnosias visuales) -astenia,anorexia,pérdida de peso , alteraciones de sueño o pérdida de libido.")
27
Fases avanzadas:-rápidamente demencia
-hipotonía -atrofia muscular -debilidad de las extremidades -sacudidas mioclónicas -atetosis Fases más avanzadas: -pérdida de funcciones físicas e intelectuales,ceguera y coma -la muerte causada por infecciones ,generalmentes pulmonares

28
-cronología de los síntomas -factores de riesgo
1.HISTORIA CLÍNICA: -cronología de los síntomas -factores de riesgo 2.PRUEBAS DE LABORATORIO: EEG:patron típico: complejos de ondas agudas y lentas (complejos trifásicos), periódicos, sincrónicos,ampliamente distribuidas en ambos hemisferios cerebrales. LCR: +normal,las proteínas suelen estar elevadas(nunca por encima de 100 mg/dl);frecuentemente aumento de la enolasa +detección de la proteína alta especificidad y sensibilidad(más de 90%);los falsos+ en pacientes con encefalitis o ictus reciente OTRAS ENFERMEDADES EN LAS QUE SE DETECTA PROTEÍNA EN LCR Encefalitis de Hashimoto Herpes simplex y otros virus Hemorragia subaracnoidea Hipoxia cerebral Encefalopatía metabólica después de intoxicación con barbitúricos Glioblastoma Encefalopatía paraneoplásica Accidente cerebrovascular agudo
, periódicos, sincrónicos,ampliamente distribuidas en ambos hemisferios cerebrales. LCR: +normal,las proteínas suelen estar elevadas(nunca por encima de 100 mg/dl);frecuentemente aumento de la enolasa. +detección de la proteína alta especificidad y sensibilidad(más de 90%);los falsos+ en pacientes con encefalitis o ictus reciente. OTRAS ENFERMEDADES EN LAS QUE SE DETECTA PROTEÍNA EN LCR. Encefalitis de Hashimoto. Herpes simplex y otros virus. Hemorragia subaracnoidea. Hipoxia cerebral. Encefalopatía metabólica después de intoxicación con barbitúricos. Glioblastoma. Encefalopatía paraneoplásica. Accidente cerebrovascular agudo.")
29
Estudio genético:más de 20 mutaciones que se traducen en cuadros clínicos diversos(E200K(Glu-Lys))
TAC: normal inicialmente,atrofia cerebral RM: imágenes de alta señal en T2 en ganglios basales DIAGNÓSTICO DEFINITIVO: biopsia o necropsia algunos casos hereditarios:análisis molecular de leucocitos de sangre periférica

30
DIAGNOSTICO POSIBLE: clínica compatible sin pruebas de laboratorio;
DIAGNOSTICO PROBABLE: clínica compatible más pruebas de laboratorio(EEG,LCR,RM); DIAGNOSTICO CONFIRMADO: clínica compatible más alteraciones neuropatológicas en biopsia o necropsia.
; DIAGNOSTICO CONFIRMADO: clínica compatible más alteraciones neuropatológicas en biopsia o necropsia.")
31
1.Demencia subaguda progresiva 2.Mioclonías
TETRADA CARACTERÍSTICA: 1.Demencia subaguda progresiva 2.Mioclonías 3.Complejos periódicos en el EEG 4.LCR normal

32
Diagnóstico diferencial:
Alzheimer (evolución más progresiva) Enf de Huntington (evolución muy progresiva 15-20años) Status epileptico no convulsivo (EEG urgente con administración de benzodiacepinas endovenosas) Ataxias cerebelosas mioclónicas progresivas
 Enf de Huntington (evolución muy progresiva 15-20años) Status epileptico no convulsivo (EEG urgente con administración de benzodiacepinas endovenosas) Ataxias cerebelosas mioclónicas progresivas.")
33
TRATAMIENTO: NO HAY TTO ESPECÍFICO Sólo tto síntomatico(aliviar los síntomas: drogas opiáceas-dolor;clonazepam,valproato de sodio-paliar el mioclono) Precauciones en el manejo de material procedente de estos pacientes(uso de guantes para sangre,LCR etc) Instrumentos desinfectados en autoclave durante una hora a 132 ºC o imersión en hipoclorito sódico al 5 % una hora La enfermedad no se transmite a través del aire o al contacto;el contacto directo o indirecto con el tejido cerebral y el líquido de la médula espinal debería evitarse 100% letalidad
 Precauciones en el manejo de material procedente de estos pacientes(uso de guantes para sangre,LCR etc) Instrumentos desinfectados en autoclave durante una hora a 132 ºC o imersión en hipoclorito sódico al 5 % una hora. La enfermedad no se transmite a través del aire o al contacto;el contacto directo o indirecto con el tejido cerebral y el líquido de la médula espinal debería evitarse. 100% letalidad.")
34
GRACIAS POR VUESTRA ATENCIÓN!!!

Presentaciones similares






>")







>")




