Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Química General II, Parte I
Grado en Química (Plan 2009) Curso: 1º Semestre: 2º © 2009 Dr. José María Fernández Álvarez
 Curso: 1º Semestre: 2º. © 2009 Dr. José María Fernández Álvarez.")
2
Química General II, Parte I
Parte I: Equilibrios y propiedades periódicas de los elementos Tema 1. Equilibrios ácido-base. Tema 2. Equilibrios de formación de complejos. Tema 3. Equilibrios heterogéneos: equilibrios de precipitación. Tema 4. Equilibrios de oxidación-reducción. Tema 5. Equilibrios de reparto. Tema 6. Propiedades periódicas de los elementos.

3
Tema 1. Equilibrios ácido-base
Electrolitos. Actividad y coeficiente de actividad Fuerza iónica. Ley límite de Debye-Hückel Concepto y escala de pH en disolventes no acuosos. Diagramas logarítmicos de concentración-pH. Disoluciones reguladoras. Capacidad reguladora. Efecto regulador en sistemas biológicos. Efecto de la constante dieléctrica del disolvente sobre el equilibrio. Equilibrios ácido-base en acético glacial y en disolventes apróticos.

4
Electrolitos Electrolito: sustancia que, en disolución, conduce la corriente eléctrica gracias a su disociación -mediante un proceso reversible- en iones libres positivos (cationes) y negativos (aniones). Fuerte: aquél que en disolución 0,1 M se encuentra casi totalmente disociado > 0,5 Electrolito Débil: aquél que en esa concentración apenas sufre disociación. < 0,5
 y negativos (aniones). Fuerte: aquél que en disolución 0,1 M se encuentra casi totalmente disociado > 0,5. Electrolito. Débil: aquél que en esa concentración apenas sufre disociación. < 0,5.")
5
Disoluciones reales a: actividad c: concentración analítica g : coeficiente de actividad a = g · c

6
3. Las interacciones entre los iones son puramente electrostáticas.
Disoluciones ideales 1. Los electrolitos fuertes están totalmente disociados en disolución acuosa a cualquier concentración. 2. Los iones se comportan como cargas puntuales creando un campo eléctrico de simetría radial. El movimiento del ión estará interferido por los iones vecinos y se crea una atmósfera iónica con carga contraria a la del ión. 3. Las interacciones entre los iones son puramente electrostáticas. 4. El coeficiente dieléctrico del medio es elevado, y puede considerarse constante (independiente de la concentración). 5. La actividad está relacionada con la concentración analítica a través del coeficiente de actividad, el cual se calcula en función de la fuerza iónica por medio de la fórmula:
. 5. La actividad está relacionada con la concentración analítica a través del coeficiente de actividad, el cual se calcula en función de la fuerza iónica por medio de la fórmula:")
7
Electrolitos monovalentes: m<0,05
Ley límite de Debye-Hückel Electrolitos monovalentes: m<0,05 Electrolitos divalentes: m <0,01 Electrolitos trivalentes: m <0,005

8
Coeficiente de actividad medio

9
a : Radio efectivo del ión solvatado
Ecuación de Debye-Hückel ampliada a : Radio efectivo del ión solvatado : Coeficiente dieléctrico del medio T: Temperatura (ºK)
")
10
25ºC: 10-13,98; 18ºC: 10-14,2; Ácido: pH < 7 pOH > 7
Concepto de pH 25ºC: 10-13,98; 18ºC: 10-14,2; Ácido: pH < 7 pOH > 7 Neutralidad: pH = 7 pOH = pH + pOH=14 Alcalino: pH> 7 pOH < 7

11
Escalas de pH Podemos representar gráficamente, para un mismo disolvente, los pares ácido-base y comparar estos valores con los de la Ka de ese mismo ácido en otro disolvente.

12
[EtOH2+]=1; [EtO-]=10-19 p EtOH2+= 0
Agua vs. Etanol [H+]=1; [OH-]=10-14 pH = 0 [H+]=10-14; [OH-]=1 pH = 14 [EtOH2+]=1; [EtO-]=10-19 p EtOH2+= 0 [EtOH2+]=10-19; [EtO-]=1 p EtOH2+= 19
![[EtOH2+]=1; [EtO-]=10-19 p EtOH2+= 0](http://slideplayer.es/slide/3290951/11/images/12/%5BEtOH2%2B%5D%3D1%3B+%5BEtO-%5D%3D10-19+%EF%83%9E+p+EtOH2%2B%3D+0.jpg "Agua vs. Etanol. [H+]=1; [OH-]=10-14 pH = 0. [H+]=10-14; [OH-]=1 pH = 14. [EtOH2+]=1; [EtO-]=10-19 p EtOH2+= 0. [EtOH2+]=10-19; [EtO-]=1 p EtOH2+= 19.")
13
14 19 HCl/Cl- (2,3) ClCH2COOH / ClCH2COO- (2,9) HAc/Ac- (4,7)
Agua vs. Etanol HCl/Cl- (2,3) ClCH2COOH / ClCH2COO- (2,9) HAc/Ac- (4,7) ClCH2COOH / ClCH2COO- (8,5) NH4+ / NH3 (9,2) HAc/Ac- (10) NH4+ / NH3 (10,4) 14 19
 ClCH2COOH / ClCH2COO- (2,9) HAc/Ac- (4,7) ClCH2COOH / ClCH2COO- (8,5) NH4+ / NH3 (9,2) HAc/Ac- (10) NH4+ / NH3 (10,4)")
14
ClCH2COOH + NH3 ClCH2COO- + NH4+
Predicción de las reacciones ácido-base ClCH2COOH + NH3 ClCH2COO- + NH4+ ClCH2COOH + HD ClCH2COO- + H2D+; k1 NH4+ + HD NH3 + H2D+; k2

15
Efecto nivelador del disolvente
Fenómeno por el cual diversos ácidos, como -por ejemplo- el HCl, HClO4 y HNO3, tienen la misma fuerza en el H2O, o el HAc y el HCl son ácidos fuertes en etilendiamina. El H2O es un disolvente NIVELADOR del HCl y HClO4, pero es NO-NIVELADOR (Diferenciador) del HCl y HAc.
 del HCl y HAc.")
16
HCl + NH2(CH2)2NH2 NH2(CH2)2NH3+ + Cl-
Efecto nivelador del disolvente HCl H2O H3O Cl- HClO H2O H3O+ + ClO4- HAc H2O H3O+ + Ac- HCl + NH2(CH2)2NH2 NH2(CH2)2NH3+ + Cl- HAc + NH2(CH2)2NH2 NH2(CH2)2NH3+ + Ac-
2NH2 NH2(CH2)2NH3+ + Cl- HAc + NH2(CH2)2NH2 NH2(CH2)2NH3+ + Ac-")
17
Efecto nivelador del disolvente
Para que un disolvente ejerza efecto nivelador sobre los ácidos debe tener una gran apetencia por los H+ (ser base fuerte o un ácido conjugado débil) Para que un disolvente tenga efecto nivelador sobre las bases ha de mostrar poca apetencia por los H+ (ser base muy débil).
 Para que un disolvente tenga efecto nivelador sobre las bases ha de mostrar poca apetencia por los H+ (ser base muy débil).")
18
HClO4 + HAc H2Ac+ + ClO4- HCl + HAc H2Ac+ + Cl-
Efecto diferenciador del disolvente HClO HAc H2Ac+ + ClO4- HCl HAc H2Ac+ + Cl- H2SO HAc H2Ac+ + HSO4-

19
Sustituyendo (1) y (2) en (3):
Cálculo del pH de un ácido (o base) fuerte (1) (2) (3) Sustituyendo (1) y (2) en (3): (4)
 fuerte. (1) (2) (3) Sustituyendo (1) y (2) en (3): (4)")
20
[H+]=ca; log[H+]=log ca; pH = -log ca; log ca= -pH
Diagrama logarítmico log ca-pH La ecuación (4): es una curva, que puede ser aproximada a 2 rectas: 1. ca >> Kw/[H+]: [H+]=ca; log[H+]=log ca; pH = -log ca; log ca= -pH Recta de pendiente -1 2. ca << Kw/[H+]: [H+]2 = Kw; log [H+]=10-7 M; pH = 7 Recta vertical que pasa por el punto pH 7
![[H+]=ca; log[H+]=log ca; pH = -log ca; log ca= -pH](http://slideplayer.es/slide/3290951/11/images/20/%5BH%2B%5D%3Dca%3B+log%5BH%2B%5D%3Dlog+ca%3B+pH+%3D+-log+ca%3B+log+ca%3D+-pH.jpg "Diagrama logarítmico log ca-pH. La ecuación (4): es una curva, que puede ser aproximada a 2 rectas: 1. ca >> Kw/[H+]: [H+]=ca; log[H+]=log ca; pH = -log ca; log ca= -pH. Recta de pendiente ca << Kw/[H+]: [H+]2 = Kw; log [H+]=10-7 M; pH = 7. Recta vertical que pasa por el punto pH 7.")
21
Diagrama logarítmico log ca-pH

22
(2) (1) (3) (4) Despejando [HA] en (1) y sustituyendo en (3):
Cálculo del pH de un ácido (o base) débil (2) (1) (3) (4) Despejando [HA] en (1) y sustituyendo en (3): Despejando [A-]:
![(2) (1) (3) (4) Despejando [HA] en (1) y sustituyendo en (3):](http://slideplayer.es/slide/3290951/11/images/22/%282%29+%281%29+%283%29+%284%29+Despejando+%5BHA%5D+en+%281%29+y+sustituyendo+en+%283%29%3A.jpg "Cálculo del pH de un ácido (o base) débil. (2) (1) (3) (4) Despejando [HA] en (1) y sustituyendo en (3): Despejando [A-]:")
23
Cálculo del pH de un ácido (o base) débil. Casos particulares
A) El ácido es extremadamente débil o está muy diluido: la concentración de protones que suministra es despreciable frente a los del disolvente. B) La acidez suministrada por el agua es muy inferior a la proporcionada por el ácido
 El ácido es extremadamente débil o está muy diluido: la concentración de protones que suministra es despreciable frente a los del disolvente. B) La acidez suministrada por el agua es muy inferior a la proporcionada por el ácido.")
24
B.1) Reacción muy desplazada
Cálculo del pH de un ácido (o base) débil. Casos particulares B.1) Reacción muy desplazada Ka >> [H+] B.2) Reacción poco desplazada Ka << [H+] Ácidos débiles y no muy diluidos B.3) Ácido parcialmente disociado y [HA][A-] No caben simplificaciones
 débil. Casos particulares. B.1) Reacción muy desplazada. Ka >> [H+] B.2) Reacción poco desplazada. Ka << [H+] Ácidos débiles y no muy diluidos. B.3) Ácido parcialmente disociado y [HA][A-] No caben simplificaciones.")
25
Grado de disociación

26
a) Predominancia del H2O:
Efecto de la dilución y de la constante. Gráfico de Flood a) Predominancia del H2O: pH = -1/2 log Ka Ca; log Ca = pKa - 2 pH b) Disociación total; H2O despreciable: pH = -log Ca; log Ca = -pH pH = 7 c) Disociación parcial; H2O despreciable:
 Predominancia del H2O: pH = -1/2 log Ka Ca; log Ca = pKa - 2 pH. b) Disociación total; H2O despreciable: pH = -log Ca; log Ca = -pH. pH = 7. c) Disociación parcial; H2O despreciable:")
27
Ácido fuerte pH pKa 4 6 log C a
Efecto de la dilución y de la constante. Gráfico de Flood pH 2 4 6 8 pKa 4 6 -2 log C a Ácido fuerte -4 -6 -8

28
Diagrama logarítmico log ca-pH
A) [H+] >> Ka; pH<<pKa , pH < pKa - 2 B) [H+] << Ka; pH>>pKa , pH > pKa + 2 C) [H+] = Ka; pH = pKa
 [H+] >> Ka; pH<<pKa , pH < pKa - 2. B) [H+] << Ka; pH>>pKa , pH > pKa + 2. C) [H+] = Ka; pH = pKa.")
29
Diagrama logarítmico log ca-pH
HA/A- con Ka = y Ca = 1,0·10-2 M

30
Diagrama logarítmico log ca-pH
HA/A- con Ka = y Ca = 1,0·10-2 M

31
Diagrama logarítmico log ca-pH
HA/A- con Ka = y Ca = 1,0·10-2 M

32
Diagrama logarítmico log ca-pH
HA/A- con Ka = y Ca = 1,0·10-2 M

33
Diagrama logarítmico log ca-pH
HA/A- con Ka = y Ca = 1,0·10-2 M

34
Punto del sistema HA/A- con Ka = 10-6 y Ca = 1,0·10-2 M
Diagrama logarítmico log ca-pH HA/A- con Ka = y Ca = 1,0·10-2 M Punto del sistema

35
Diagrama logarítmico log ca-pH
0,30

36
Diagrama logarítmico log ca-pH

37
Diagrama logarítmico log ca-pH

38
Diagrama logarítmico log ca-pH

39
Diagrama logarítmico log ca-pH

40
Diagrama logarítmico log ca-pH

41
Diagrama logarítmico log ca-pH

42
Diagrama logarítmico log ca-pH

43
Grado de disociación y fracción molar.

44
Ejemplos HA de pKa = 5,0 y Ca = 1,0·10-2 M, calcule:
a) [HA], [A-], pH; b) xHA, xA-; c) Ejemplos
 [HA], [A-], pH; b) xHA, xA-; c) Ejemplos.")
45
Conocido pH = 3,0 (disociación de HA puro) y pK = 5,0, calcule Ca
Ejemplos

46
Ejemplos HA de pKa = 10,0 y Ca = 1,0·10-1 M, calcule:
a) [HA], [A-], pH; b) xHA, xA-; c) Ejemplos
 [HA], [A-], pH; b) xHA, xA-; c) Ejemplos.")
47
Hidrólisis de una sal de ácido débil y base fuerte

48
Hidrólisis de una sal de ácido débil y base fuerte
Si despreciamos el H2O, y la hidrólisis no es muy grande (Kh<<[OH-]): Hidrólisis de una sal de ácido fuerte y base débil Si despreciamos el H2O, y la hidrólisis no es muy grande (Kh<<[H+]):
: Hidrólisis de una sal de ácido fuerte y base débil. Si despreciamos el H2O, y la hidrólisis no es muy grande (Kh<<[H+]):")
49
Simplificaciones: Hidrólisis. Ecuación de Noyes
Simplificaciones:

50
A) sal de ácido débil y base fuerte: ácido débil: Ka finito
Hidrólisis. Ecuación de Noyes A) sal de ácido débil y base fuerte: ácido débil: Ka finito base fuerte: Kb’ infinito B) sal de ácido fuerte y base débil: ácido fuerte: Ka infinito base débil: Kb’ finito
 sal de ácido débil y base fuerte: ácido débil: Ka finito. base fuerte: Kb’ infinito. B) sal de ácido fuerte y base débil: ácido fuerte: Ka infinito. base débil: Kb’ finito.")
51
Su pH permanece invariable a la dilución.
Disoluciones reguladoras (amortiguadoras , tampón, buffer) Su pH permanece invariable a la dilución. Admiten cantidades moderadas de ácido o de base, sin que apenas varíe su pH. Tipos: Ácidos o bases fuertes moderadamente concentrados. (HCl, NaOH) Ácido débil y un exceso de base conjugada; base débil y un exceso de su ácido conjugado. (HAc-Ac-; NH4OH-NH4Cl) 10 mL HCl 0,1 M pH = 1 Diluyendo a 1 L: HCl 0,001 M pH = 3 1 L H2O pH = 7 + 1 mL de HCl 0,1 M: [H+]= 10-4 M pH = 4 100 mL HCl 0,5 M pH = 0,301 + 1 mL NaOH 0,1 M: [H+]=4,94·10-1M pH = 0,306
 Su pH permanece invariable a la dilución. Admiten cantidades moderadas de ácido o de base, sin que apenas varíe su pH. Tipos: Ácidos o bases fuertes moderadamente concentrados. (HCl, NaOH) Ácido débil y un exceso de base conjugada; base débil y un exceso de su ácido conjugado. (HAc-Ac-; NH4OH-NH4Cl) 10 mL HCl 0,1 M pH = 1. Diluyendo a 1 L: HCl 0,001 M. pH = 3. 1 L H2O pH = mL de HCl 0,1 M: [H+]= 10-4 M. pH = mL HCl 0,5 M pH = 0, mL NaOH 0,1 M: [H+]=4,94·10-1M. pH = 0,306.")
52
Reguladora HAc/NaAc: reserva alcalina reserva ácida NaAc Na+ + Ac-
Disoluciones reguladoras: reservas ácida y alcalina Reguladora HAc/NaAc: NaAc Na+ + Ac- HAc Ac- + H+ H+ + Ac- HAc OH- + HAc Ac- + H2O reserva alcalina reserva ácida Para que el pH se modifique en una unidad, es preciso que la proporción de HAc/Ac- varíe en un factor de 10.

53
HA A- + H+ pKa =4,00 0,1-x x x A- + H2O HA + OH- pKb =10,00 0,1-x
Disoluciones reguladoras Las concentraciones de un ácido débil y de su base conjugada permanecen prácticamente inalteradas cuando se mezclan. HA A H+ pKa =4,00 0,1-x x x A H2O HA + OH- pKb =10,00 0,1-x x x

54
Disoluciones reguladoras
+ 12,0 mL de HCl 1,0 M:

55
Disoluciones reguladoras
+ 12,0 mL de HCl 1,0 M:

56
Disoluciones reguladoras
+ 12,0 mL de HCl 1,0 M:

57
Coeficientes de actividad:
Disoluciones reguladoras: limitaciones Coeficientes de actividad: Temperatura: pKa = f (T) Disoluciones diluidas o valores extremos de pH: HA H+ + A- Ka A- + H2O HA + OH- Kb
 Disoluciones diluidas o valores extremos de pH: HA H+ + A- Ka. A- + H2O HA + OH- Kb.")
58
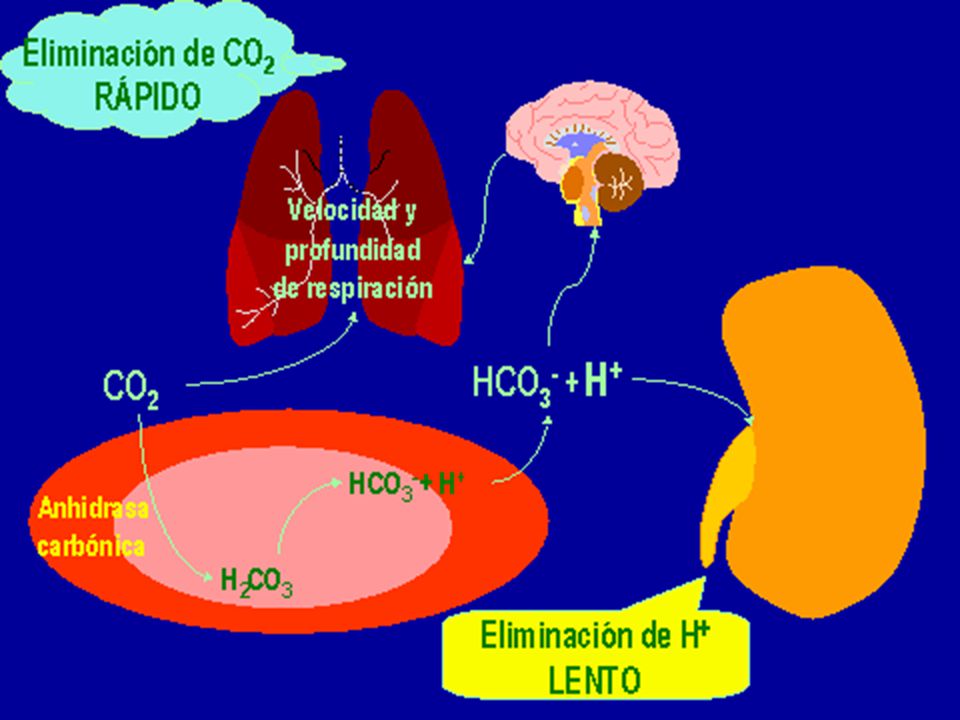
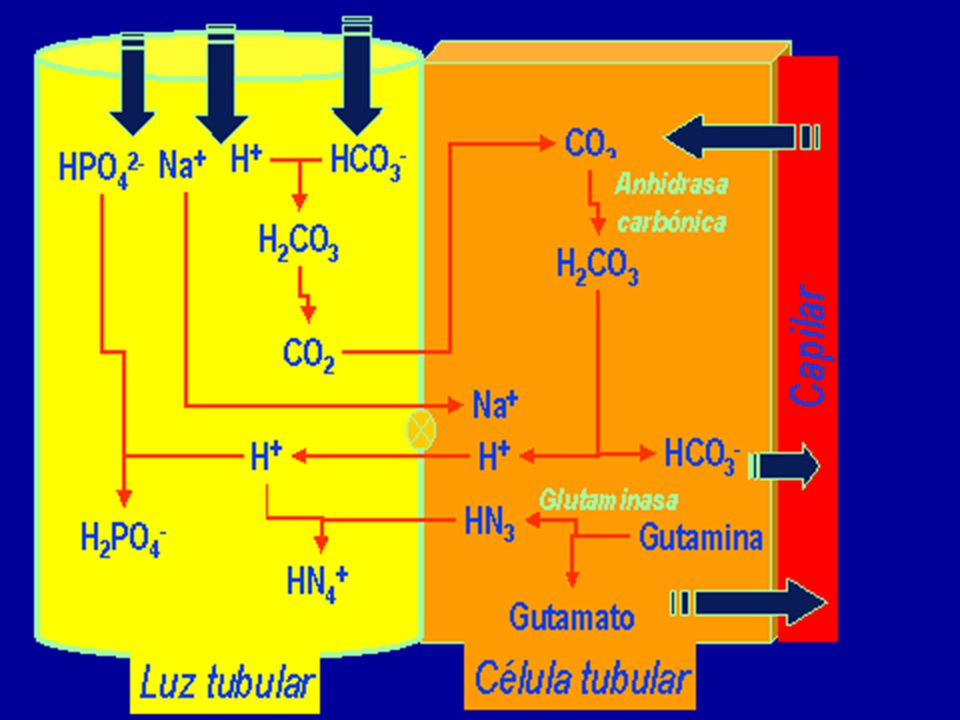
H2O + CO2(ac) + Hb-(ac) HHb(ac) + HCO3-
Disoluciones reguladoras fisiológicas Pulmones: 30 L ácido 1 M; Riñones: 100 mL ácido 1 M* Amortiguadores en sangre: proteinas, bicarbonato, fosfatos, hemoglobina (HHb) y oxihemoglobina (HHbO2) H2O + CO2(ac) + Hb-(ac) HHb(ac) + HCO3- Metabolismo en tejidos A los pulmones pKa (H2CO3 ;6,1) < pKa (HHb;7,93) HCO3-(ac) + HHbO2(ac) HbO2-(ac) + H2O + CO2(g) pKa (HHbO2) = 6,68 A los tejidos Exhalado
 y oxihemoglobina (HHbO2) H2O + CO2(ac) + Hb-(ac) HHb(ac) + HCO3- Metabolismo en tejidos. A los pulmones. pKa (H2CO3 ;6,1) < pKa (HHb;7,93) HCO3-(ac) + HHbO2(ac) HbO2-(ac) + H2O + CO2(g) pKa (HHbO2) = 6,68. A los tejidos. Exhalado.")
61
Constantes sucesivas y globales de formación. Reacciones parásitas.
Tema 2. Equilibrios de formación de complejos Reacciones de formación de complejos y tipos de complejos de interés analítico. Constantes sucesivas y globales de formación. Reacciones parásitas. Constantes condicionales. Reacciones de desplazamiento y enmascaramiento.

62
Complejo: átomo o ión central (ácido), normalmente metálico, rodeado por un grupo de iones o moléculas (ligandos, básicos) unidos a él por enlaces coordinados. Enlace covalente coordinado (semipolar o dativo): compartición de un par electrónico que procede, íntegramente, de un sólo átomo de los que se enlazan. Número de coordinación: nº de enlaces (pares electrónicos) que pueden incidir sobre el ión central. Las propiedades de los iones metálicos quedan atenuadas, apareciendo las características típicas de la nueva especie: el complejo.
: compartición de un par electrónico que procede, íntegramente, de un sólo átomo de los que se enlazan. Número de coordinación: nº de enlaces (pares electrónicos) que pueden incidir sobre el ión central. Las propiedades de los iones metálicos quedan atenuadas, apareciendo las características típicas de la nueva especie: el complejo.")
63
Complejos de utilidad analítica
Atendiendo a la naturaleza del ligando, los complejos se pueden clasificar en: Monodentados Polidentados Ligandos monodentados: son aquellos que participan en la formación de compuestos de coordinación con un enlace por molécula de ligando: H2O, NH3, F-, CN- (un sólo átomo donador). Ligandos polidentados: son los que participan en la formación de compuestos de coordinación con 2 ó más enlaces con el ión metálico por molécula de ligando. Pueden ser bidentados, tridentados, tetradentados, etc.
. Ligandos polidentados: son los que participan en la formación de compuestos de coordinación con 2 ó más enlaces con el ión metálico por molécula de ligando. Pueden ser bidentados, tridentados, tetradentados, etc.")
64
Complejos con ligandos monodentados

65
Complejos con ligandos polidentados: Quelatos

66
En muchos casos: KML > KML2 > KMLn
Labilidad de los complejos Independientemente de la estabilidad de los complejos, que viene dada por su correspondiente constante, un complejo se dice LÁBIL si intercambia los ligandos rápidamente; se dice INERTE si intercambia los ligandos con lentitud. Constantes sucesivas de formación M + L ML M L + L ML2 y, en general: MLn L MLn En muchos casos: KML > KML2 > KMLn

67
Constantes globales de formación
M L ML2 Especies en disolución

68
Especies en disolución

69
Efecto de la concentración de L

70
Efecto de la concentración de L
Para [NH3]= 10-1 M:

71
Efecto de la concentración de L

72
Número promedio de grupos coordinados por ión metálico presente.
Índice de coordinación medio Número promedio de grupos coordinados por ión metálico presente. Para complejos MONONUCLEARES, depende sólo de [L], y no de la concentración total de metal.

73
Índice de coordinación medio: efecto de [L]
![Índice de coordinación medio: efecto de [L]](http://slideplayer.es/slide/3290951/11/images/73/%C3%8Dndice+de+coordinaci%C3%B3n+medio%3A+efecto+de+%5BL%5D.jpg "Índice de coordinación medio: efecto de [L]")
74
Complejos polinucleares
m M + n L MmLn

75
Zn2+ + Y4- ZnY2- Reacciones secundarias, laterales o parásitas:
Constantes condicionales Zn Y ZnY2- Reacciones secundarias, laterales o parásitas:

76
Constantes condicionales
M + L ML [M’]: suma de las concentraciones de todas las especies formadas en las que M no se ha combinado con L. [Zn’] = [Zn2+] + [Zn(NH3)2+] +…+ [Zn(OH)+] [L’]: suma de las concentraciones del ligando en todas sus formas excepto aquellas en las que se ha combinado con M.
2+] +…+ [Zn(OH)+] [L’]: suma de las concentraciones del ligando en todas sus formas excepto aquellas en las que se ha combinado con M.")
77
Coeficientes de Schwarzenbach
1 = 1: ausencia de reacciones secundarias M + L ML m M + n L MmLn

78
En presencia de un ligando interferente A que compite con L:
Coeficientes de Schwarenbach En presencia de un ligando interferente A que compite con L: En presencia de un catión interferente B que compite con M :

79
Equilibrios concurrentes
A) Fuerza iónica Un aumento de , implica una disminución de 2 y, por tanto, KML también decrece.
 Fuerza iónica. Un aumento de , implica una disminución de 2 y, por tanto, KML también decrece.")
80
B) Efecto del pH L + H+ HL+ M + L ML HL+ L + H+
Equilibrios concurrentes B) Efecto del pH L + H HL+ M + L ML HL+ L + H+ La reacción de disociación del complejo es: ML + H+ M + HL+ Suponiendo que CM = CL: En el equilibrio: Si asumimos que el complejo es bastante perfecto, podemos considerar que [ML]CM:
 Efecto del pH. L + H+ HL+ M + L ML. HL+ L + H+ La reacción de disociación del complejo es: ML + H+ M + HL+ Suponiendo que CM = CL: En el equilibrio: Si asumimos que el complejo es bastante perfecto, podemos considerar que [ML]CM:")
81
En general, para un complejo MLn:
Equilibrios concurrentes En general, para un complejo MLn: Y para un complejo MmLn: Simplificaciones relevantes A) Si pH > pKa+1, [H+]/Ka < 0,1 : La disociación del complejo resulta independiente del pH B) Si pH < pKa-1, [H+]/Ka > 10 : Ej.:Fe L FeL Kf =105,5 L H+ HL Ka =10-3,2
 Si pH > pKa+1, [H+]/Ka < 0,1 : La disociación del complejo resulta independiente del pH. B) Si pH < pKa-1, [H+]/Ka > 10 : Ej.:Fe3+ + L FeL3+ Kf =105,5. L + H+ HL+ Ka =10-3,2.")
82
ML M + L C) Dilución Co(1-) Co Co
Equilibrios concurrentes C) Dilución ML M + L Co(1-) Co Co Si se diluye lo suficiente, puede destruirse el complejo en su totalidad.
 Dilución. ML M + L. Co(1-) Co Co Si se diluye lo suficiente, puede destruirse el complejo en su totalidad.")
83
Ejemplo: Estudio de la variación de la disociación de los complejos del Hg2+ con SCN-
La dilución fuerza la desaparición sucesiva de Hg(SCN)42- y Hg(SCN)3-; el Hg(SCN)+, mucho más estable, resiste más a la dilución.
42- y Hg(SCN)3-; el Hg(SCN)+, mucho más estable, resiste más a la dilución.")
84
Donador Aceptor + P Complejo Catión Ligando
Reacciones de desplazamiento Donador Aceptor P Complejo Catión Ligando

85
Kd Estabilidad Kf DONADORES FUERTES ACEPTORES FUERTES
Reacciones de desplazamiento: predicción de las reacciones DONADORES FUERTES Kd Estabilidad Kf ACEPTORES FUERTES

86
Reacciones de desplazamiento: predicción de las reacciones
MY M + Y4-

87
En un equilibrio único: ML M + L
Cálculo de [L] En un equilibrio único: ML M + L En una reguladora de Y4-: CaY2-/Ca2+ En reacciones de desplazamiento:

88
a) Complejos característicos aptos para reacciones analíticas
Aplicaciones analíticas a) Complejos característicos aptos para reacciones analíticas Identificaciones Con ligandos monodentados Con ligandos polidentados Determinaciones Separaciones Complexonas b) Reacciones de enmascaramiento y desenmascaramiento c) Variación de la capacidad reactiva de algunos iones Modificación de la fuerza de los ácidos débiles Modificación de los potenciales red-ox de los sistemas
 Complejos característicos aptos para reacciones analíticas. Identificaciones. Con ligandos monodentados. Con ligandos polidentados. Determinaciones. Separaciones. Complexonas. b) Reacciones de enmascaramiento y desenmascaramiento. c) Variación de la capacidad reactiva de algunos iones. Modificación de la fuerza de los ácidos débiles. Modificación de los potenciales red-ox de los sistemas.")
89
A) Con ligandos monodentados:
Aplicaciones analíticas: identificaciones A) Con ligandos monodentados: Fe(SCN)2+ Rojo intenso; Cu(NH3)42+ Azul intenso B) Con ligandos polidentados:
 Con ligandos monodentados: Fe(SCN)2+ Rojo intenso; Cu(NH3)42+ Azul intenso. B) Con ligandos polidentados:")
90
* * negro amarillo rojo-naranja alcaloide Con ligandos monodentados:
Aplicaciones analíticas: determinaciones Con ligandos monodentados: Ag(CN)2- : Cianometrías Con ligandos polidentados: Ca2+, Mg2+ con AEDT : Quelatometrías Aplicaciones analíticas: separaciones * alcaloide negro amarillo rojo-naranja *
2- : Cianometrías. Con ligandos polidentados: Ca2+, Mg2+ con AEDT : Quelatometrías. Aplicaciones analíticas: separaciones. * alcaloide. negro. amarillo. rojo-naranja. *")
91
* * Ni(II)-DMG: rojo rosado
Aplicaciones analíticas: enmascaramiento * Ni(II)-DMG: rojo rosado Fe(II)-DMG: rojo: interfiere. Se enmascara con ácido tartárico *
-DMG: rojo rosado. Fe(II)-DMG: rojo: interfiere. Se enmascara con ácido tartárico. *")
92
* * Modificación de la fuerza de los ácidos:
Aplicaciones analíticas: desenmascaramiento C. oxidantes Aplicaciones analíticas: variación de la reactividad * Modificación de la fuerza de los ácidos: * Modificación de potenciales red-ox Disolución de metales nobles

93
Solubilidad y producto de solubilidad. Precipitación fraccionada.
Tema 3. Equilibrios heterogéneos: precipitación Solubilidad y producto de solubilidad. Precipitación fraccionada. Influencia de las reacciones ácido-base y de formación de complejos sobre la precipitación. Solubilización de precipitados. Precipitaciones inducidas.

94
B) [AB]dis es constante: [AB]dis=S0
Solubilidad A) El equilibrio es independiente de la cantidad de sólido presente en contacto con la disolución. B) [AB]dis es constante: [AB]dis=S0 1. [AB]dis < S0: tenderá a disolverse más ABpdo. La disolución no estaba saturada. 2. [AB]dis > S0: tenderá a precipitar parte de ABdis. La disolución estaba sobresaturada. 3. [AB]dis = S0: situación de equilibrio. La disolución está saturada. [AB]dis = SOLUBILIDAD
![B) [AB]dis es constante: [AB]dis=S0](http://slideplayer.es/slide/3290951/11/images/94/B%29+%5BAB%5Ddis+es+constante%3A+%5BAB%5Ddis%3DS0.jpg "Solubilidad. A) El equilibrio es independiente de la cantidad de sólido presente en contacto con la disolución. B) [AB]dis es constante: [AB]dis=S0. 1. [AB]dis < S0: tenderá a disolverse más ABpdo. La disolución no estaba saturada. 2. [AB]dis > S0: tenderá a precipitar parte de ABdis. La disolución estaba sobresaturada. 3. [AB]dis = S0: situación de equilibrio. La disolución está saturada. [AB]dis = SOLUBILIDAD.")
95
Solubilidad y Producto de solubilidad
Eq. heterogéneo Eq. homogéneo Para una estequiometría general

96
S = [AB]dis + [A-] = [AB]dis + [B+]
Solubilidad aparente Solubilidad aparente es la suma de la parte de sustancia que está disuelta ( e indisociada) más la que está en forma iónica. S = [AB]dis + [A-] = [AB]dis + [B+] Ejemplo: Cd(OH) Cd OH-
![S = [AB]dis + [A-] = [AB]dis + [B+]](http://slideplayer.es/slide/3290951/11/images/96/S+%3D+%5BAB%5Ddis+%2B+%5BA-%5D+%3D+%5BAB%5Ddis+%2B+%5BB%2B%5D.jpg "Solubilidad aparente. Solubilidad aparente es la suma de la parte de sustancia que está disuelta ( e indisociada) más la que está en forma iónica. S = [AB]dis + [A-] = [AB]dis + [B+] Ejemplo: Cd(OH)2 Cd OH-")
97
A) Para un electrolito indisociado: S = S0
Solubilidad aparente A) Para un electrolito indisociado: S = S0 B) Electrolito fuerte: K > S0
 Para un electrolito indisociado: S = S0. B) Electrolito fuerte: K > S0.")
98
Ejemplos: AgCl; m=n=1 BaCl2; m=1, n=2 Bi2S3; m=3, n=2
Relación entre Ps y S en agua pura Ejemplos: AgCl; m=n=1 BaCl2; m=1, n=2 Bi2S3; m=3, n=2 NH4MgAsO4; m=1, n=1, p=1

99
The molar solubility depends on the stoichiometry of the salt.
Relación entre Ps y S en agua pura The molar solubility depends on the stoichiometry of the salt. A 1:1 salt is less soluble than a nonsymmetric salt with the same Ksp. ©Gary Christian, Analytical Chemistry, 6th Ed. (Wiley)
")
100
Relación entre Ps y S: consecuencias
1) Para que una disolución iónica precipite es necesario que el producto de las concentraciones de los iones formados en su disociación iónica elevados a sus respectivos coeficientes sea mayor que el producto de solubilidad: Pi > Ps 2) Una sustancia poco soluble se disolverá siempre que dicho producto sea inferior al Ps: Pi < Ps 3) Si ese producto es igual al Ps tendremos una disolución saturada de esa sustancia: Pi = Ps
 Para que una disolución iónica precipite es necesario que el producto de las concentraciones de los iones formados en su disociación iónica elevados a sus respectivos coeficientes sea mayor que el producto de solubilidad: Pi > Ps. 2) Una sustancia poco soluble se disolverá siempre que dicho producto sea inferior al Ps: Pi < Ps. 3) Si ese producto es igual al Ps tendremos una disolución saturada de esa sustancia: Pi = Ps.")
101
Relación entre Ps y S: consecuencias
1) Para que una disolución iónica precipite es necesario que el producto de las concentraciones de los iones formados en su disociación iónica elevados a sus respectivos coeficientes sea mayor que el producto de solubilidad: Pi > Ps 2) Una sustancia poco soluble se disolverá siempre que dicho producto sea inferior al Ps: Pi < Ps 3) Si ese producto es igual al Ps tendremos una disolución saturada de esa sustancia: Pi = Ps
 Para que una disolución iónica precipite es necesario que el producto de las concentraciones de los iones formados en su disociación iónica elevados a sus respectivos coeficientes sea mayor que el producto de solubilidad: Pi > Ps. 2) Una sustancia poco soluble se disolverá siempre que dicho producto sea inferior al Ps: Pi < Ps. 3) Si ese producto es igual al Ps tendremos una disolución saturada de esa sustancia: Pi = Ps.")
102
Relación entre Ps y S: consecuencias
1) Para que una disolución iónica precipite es necesario que el producto de las concentraciones de los iones formados en su disociación iónica elevados a sus respectivos coeficientes sea mayor que el producto de solubilidad: Pi > Ps 2) Una sustancia poco soluble se disolverá siempre que dicho producto sea inferior al Ps: Pi < Ps 3) Si ese producto es igual al Ps tendremos una disolución saturada de esa sustancia: Pi = Ps
 Para que una disolución iónica precipite es necesario que el producto de las concentraciones de los iones formados en su disociación iónica elevados a sus respectivos coeficientes sea mayor que el producto de solubilidad: Pi > Ps. 2) Una sustancia poco soluble se disolverá siempre que dicho producto sea inferior al Ps: Pi < Ps. 3) Si ese producto es igual al Ps tendremos una disolución saturada de esa sustancia: Pi = Ps.")
103
Relación entre Ps y S: consecuencias
1) Para que una disolución iónica precipite es necesario que el producto de las concentraciones de los iones formados en su disociación iónica elevados a sus respectivos coeficientes sea mayor que el producto de solubilidad: Pi > Ps 2) Una sustancia poco soluble se disolverá siempre que dicho producto sea inferior al Ps: Pi < Ps 3) Si ese producto es igual al Ps tendremos una disolución saturada de esa sustancia: Pi = Ps
 Para que una disolución iónica precipite es necesario que el producto de las concentraciones de los iones formados en su disociación iónica elevados a sus respectivos coeficientes sea mayor que el producto de solubilidad: Pi > Ps. 2) Una sustancia poco soluble se disolverá siempre que dicho producto sea inferior al Ps: Pi < Ps. 3) Si ese producto es igual al Ps tendremos una disolución saturada de esa sustancia: Pi = Ps.")
104
Precipitación fraccionada
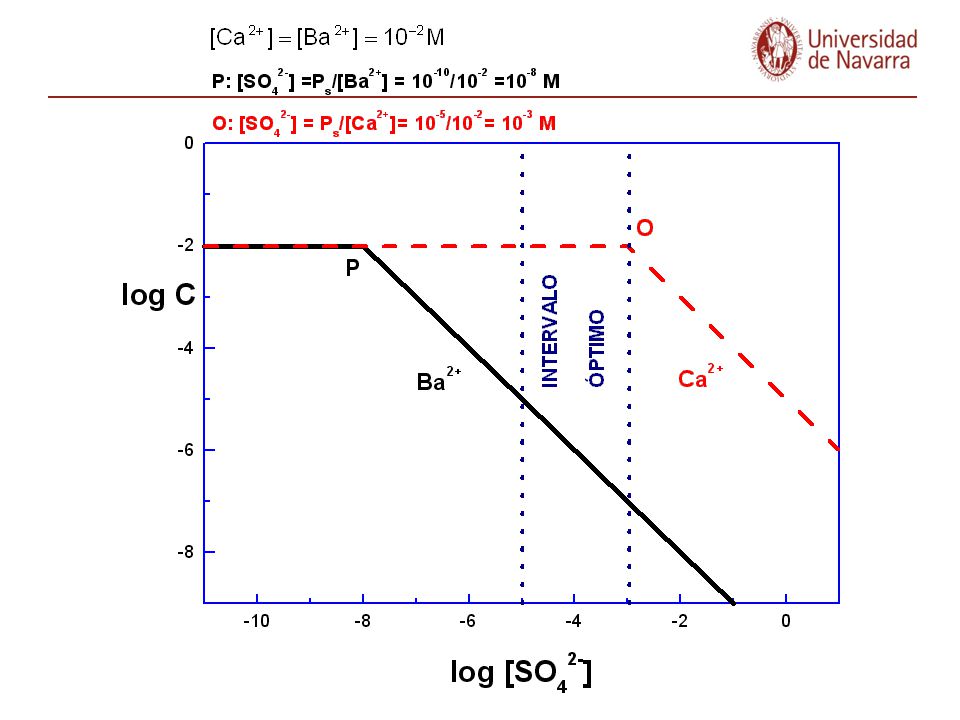
Al añadir C sobre una disolución que contiene A y B, precipitará antes aquél que necesite menor [C] para que el producto iónico supere el Ps.

105
[A]final = [A]inicial ·10-3
Precipitación fraccionada: ejemplos Convenio: Una especie está totalmente precipitada cuando su concentración en disolución es el 0,1% de la inicial, es decir: [A]final = [A]inicial ·10-3 El reactivo precipitante necesario para comenzar las precipitaciones respectivas es: Cuando [Ag+] añadida llegue a 10-6 M, habrá desaparecido cuantitativamente el Cl- de la disolución y todo el CrO42- permanecerá disuelto, pues precisa de una [Ag+]=6,3·10-6 M para empezar a precipitar. Por tanto el Cl- precipita antes y, además, su precipitación habrá terminado cuando aún no haya comenzado a precipitar nada de CrO42-, Precipitación fraccionada.
![[A]final = [A]inicial ·10-3](http://slideplayer.es/slide/3290951/11/images/105/%5BA%5Dfinal+%3D+%5BA%5Dinicial+%C2%B710-3.jpg "Precipitación fraccionada: ejemplos. Convenio: Una especie está totalmente precipitada cuando su concentración en disolución es el 0,1% de la inicial, es decir: [A]final = [A]inicial ·10-3. El reactivo precipitante necesario para comenzar las precipitaciones respectivas es: Cuando [Ag+] añadida llegue a 10-6 M, habrá desaparecido cuantitativamente el Cl- de la disolución y todo el CrO42- permanecerá disuelto, pues precisa de una [Ag+]=6,3·10-6 M para empezar a precipitar. Por tanto el Cl- precipita antes y, además, su precipitación habrá terminado cuando aún no haya comenzado a precipitar nada de CrO42-, Precipitación fraccionada.")
106
Precipitación fraccionada: ejemplos
[Br-] [Ag+]

107
Precipitación fraccionada: ejemplos
En esta situación no se logra la precipitación fraccionada del Cl-, pues antes de rebajar su concentración al 0,1% de su valor inicial ha comenzado a precipitar el cromato.

108
Precipitación fraccionada: MÉTODOS GRÁFICOS

109
Precipitación fraccionada: MÉTODOS GRÁFICOS

110
Precipitación fraccionada: MÉTODOS GRÁFICOS

112
Precipitación simultánea
Cuando varios iones comienzan a precipitar simultáneamente con un ión común, puede determinarse la proporción en que dichos iones se encuentran en disolución.

113
2) Naturaleza del disolvente
Solubilidad de los precipitados 1) Temperatura La disolución de una sustancia suele ser un proceso endotérmico. Ejemplo: Ps AgCl 10ºC: 3,7·10-11 20ºC: 1,6·10-10 50ºC: 1,3·10-9 Aplicación: solubilización de halogenuros: PbCl2 2) Naturaleza del disolvente Una disminución de la constante dieléctrica suele provocar una solubilidad menor.
 Temperatura. La disolución de una sustancia suele ser un proceso endotérmico. Ejemplo: Ps AgCl 10ºC: 3,7· ºC: 1,6· ºC: 1,3·10-9. Aplicación: solubilización de halogenuros: PbCl2. 2) Naturaleza del disolvente. Una disminución de la constante dieléctrica suele provocar una solubilidad menor.")
114
3) Factores cristaloquímicos o morfológicos
Solubilidad de los precipitados 3) Factores cristaloquímicos o morfológicos Tamaño de partícula: a menor tamaño de partícula mayor solubilidad. Grado de hidratación: la solubilidad es mayor en los compuestos hidratados. Envejecimiento: la solubilidad disminuye con el tiempo.
 Factores cristaloquímicos o morfológicos. Tamaño de partícula: a menor tamaño de partícula mayor solubilidad. Grado de hidratación: la solubilidad es mayor en los compuestos hidratados. Envejecimiento: la solubilidad disminuye con el tiempo.")
115
4) Presencia de electrolitos
Solubilidad de los precipitados 4) Presencia de electrolitos Electrolito inerte: la solubilidad aumenta con la concentración y la carga del electrolito inerte. Electrolito no inerte (efecto del ión común):
 Presencia de electrolitos. Electrolito inerte: la solubilidad aumenta con la concentración y la carga del electrolito inerte. Electrolito no inerte (efecto del ión común):")
116
Solubilidad de los precipitados
The common ion effect is used to decrease the solubility. Sulfate concentration is the amount in equilibrium and is equal to the BaSO4 solubility. In absence of excess barium ion, solubility is 10-5 M. ©Gary Christian, Analytical Chemistry, 6th Ed. (Wiley) Fig. Predicted effect of excess barium ion on solubility of BaSO4.
 Fig. Predicted effect of excess barium ion on solubility of BaSO4.")
117
Solubilidad de los precipitados
La adición de KNO3 0,05 M sobre una disolución saturada de Hg2(IO3)2, provoca la disolución de más sal hasta alcanzar un valor de [Hg22+]= 1,0·10-6 M. - +
2, provoca la disolución de más sal hasta alcanzar un valor de [Hg22+]= 1,0·10-6 M. - +")
118
PS = PS0/Ag+SO42- Solubilidad de los precipitados
Solubility increases with increasing ionic strength as activity coefficients decrease. ©Gary Christian, Analytical Chemistry, 6th Ed. (Wiley) Predicted effect of increased ionic strength on solubility of BaSO4. Solubility at zero ionic strength is 1.0 x 10-5 M.
 Predicted effect of increased ionic strength on solubility of. BaSO4. Solubility at zero ionic strength is 1.0 x 10-5 M.")
119
5) Otras reacciones simultáneas
Solubilidad de los precipitados 5) Otras reacciones simultáneas Si, simultáneamente, tiene lugar la reacción: Se disolverá más AB, para que siga cumpliéndose el Ps.
 Otras reacciones simultáneas. Si, simultáneamente, tiene lugar la reacción: Se disolverá más AB, para que siga cumpliéndose el Ps.")
120
Solubilidad condicional
Reacción secundaria o parásita: a) S’ aumenta con la [C] b) S’ aumenta al disminuir Kd o al aumentar Kf, es decir, cuanto mayor sea la tendencia a producirse la reacción secundaria.
 S’ aumenta con la [C] b) S’ aumenta al disminuir Kd o al aumentar Kf, es decir, cuanto mayor sea la tendencia a producirse la reacción secundaria.")
121
Si tanto A como B experimentan reacción secundaria:
Recordatorio: coeficiente de Swarcenbach Luego: Si tanto A como B experimentan reacción secundaria: Para un compuesto AmBn Si A y B experimentan reacciones secundarias:

122
HA HAdis A- + H+ S0= cte. A) pH << pKa S’ = S0 = cte.
Reacción secundaria: la sustancia poco soluble es un ácido débil HA HAdis A- + H+ S0= cte. A) pH << pKa S’ = S0 = cte. B) pH >> pKa
 pH << pKa S’ = S0 = cte. B) pH >> pKa ")
123
BOH BOHdis B+ + OH- S0 = cte. A) pH << pKa
Reacción secundaria: la sustancia poco soluble es una base débil BOH BOHdis B+ + OH- S0 = cte. A) pH << pKa B) pH >> pKa S’=S0 = cte.
 pH << pKa B) pH >> pKa S’=S0 = cte.")
124
MO- + H+ como ácido MOH MOHdis M+ + OH- como base
Reacción secundaria: la sustancia poco soluble es un anfolito MO- + H+ como ácido MOH MOHdis M+ + OH- como base A) pH << pK1: S’ K’ [H+]; B) pH >> pK2: S’ K/ [H+]
 pH << pK1: S’ K’ [H+]; B) pH >> pK2: S’ K/ [H+]")
125
Ejemplo: [Al3+]=10-2 M AlO H3O+ ácido Al(OH)3 Al(OH)3, dis Al OH base
![Ejemplo: [Al3+]=10-2 M AlO2- + H3O+ ácido. Al(OH)3 Al(OH)3, dis.](http://slideplayer.es/slide/3290951/11/images/125/Ejemplo%3A+%5BAl3%2B%5D%3D10-2+M+AlO2-+%2B+H3O%2B+%C3%A1cido.+%EF%82%AF+Al%28OH%293+%EF%81%84+Al%28OH%293%2C+dis..jpg "Al OH- base.")
126
A pH=4, todo el Al(OH)3 está como Al3+.
Simplificaciones: 1) A pH=4, todo el Al(OH)3 está como Al3+.
 A pH=4, todo el Al(OH)3 está como Al3+.")
127
2) A pH=10, todo el Al(OH)3 está como AlO2-.
 A pH=10, todo el Al(OH)3 está como AlO2-.")
128
Precipitación: Pi > Ps
Solubilización de precipitados Precipitación: Pi > Ps Solubilización: Pi<Ps Adición de iones inertes Aumento de la Tª Incremento del Ps Ácido-base Complejación Red-ox Formación de compuestos más insolubles Descenso de las concentraciones iónicas

129
A) Reacciones ácido-base 1) Formación de ácido débil:
Solubilización de precipitados A) Reacciones ácido-base 1) Formación de ácido débil: Todos los compuestos cuyo anión sea básico se disolverán en ácidos fuertes por disminuir la concentración del anión por formación del ácido débil correspondiente, aumentando así la S’ y el Ps’. Ej.: BaCO3, Ps 10-9 se disuelve en ácidos BaSO4, Ps 10-10 no se disuelve en ácidos
 Reacciones ácido-base. 1) Formación de ácido débil: Todos los compuestos cuyo anión sea básico se disolverán en ácidos fuertes por disminuir la concentración del anión por formación del ácido débil correspondiente, aumentando así la S’ y el Ps’. Ej.: BaCO3, Ps 10-9 se disuelve en ácidos. BaSO4, Ps 10-10 no se disuelve en ácidos.")
130
pH=2 pH=10

131
A) Reacciones ácido-base 2) Formación de base débil:
Solubilización de precipitados A) Reacciones ácido-base 2) Formación de base débil: Algunos hidróxidos se disuelven en las sales amónicas originadas por la formación de la correspondiente base débil poco disociada: NH4+ + OH- NH3 + H2O Ej.: Disolución de Mg2+ que precipita por adición de OH-: Mg OH- Mg(OH)2; Se disolverá en medio amoniacal.
 Reacciones ácido-base. 2) Formación de base débil: Algunos hidróxidos se disuelven en las sales amónicas originadas por la formación de la correspondiente base débil poco disociada: NH4+ + OH- NH3 + H2O. Ej.: Disolución de Mg2+ que precipita por adición de OH-: Mg OH- Mg(OH)2; Se disolverá en medio amoniacal.")
132
B) Formación de complejos poco disociados
Solubilización de precipitados B) Formación de complejos poco disociados La disolución dependerá del valor relativo de la constante de estabilidad del ión complejo respecto del correspondiente Ps.
 Formación de complejos poco disociados. La disolución dependerá del valor relativo de la constante de estabilidad del ión complejo respecto del correspondiente Ps.")
133
Efecto del LIGANDO [NH3]total = 0,22+2·10-2 = 0,24 M
![Efecto del LIGANDO [NH3]total = 0,22+2·10-2 = 0,24 M](http://slideplayer.es/slide/3290951/11/images/133/Efecto+del+LIGANDO+%5BNH3%5Dtotal+%3D+0%2C22%2B2%C2%B710-2+%3D+0%2C24+M.jpg "Efecto del LIGANDO [NH3]total = 0,22+2·10-2 = 0,24 M")
134
Efecto del PS

135
AgI (Ps=10-16,1): S’=2,8·10-4 M en [NH3]=10 M
Efecto de la estabilidad del complejo formado Cuanto más perfecto sea el complejo formado, tanto más fácilmente se disolverá el precipitado. AgI (Ps=10-16,1): S’=2,8·10-4 M en [NH3]=10 M Se recurre a un complejo más perfecto (Kd menor) Para [CN-]=10 M:
![AgI (Ps=10-16,1): S’=2,8·10-4 M en [NH3]=10 M](http://slideplayer.es/slide/3290951/11/images/135/AgI+%28Ps%3D10-16%2C1%29%3A+S%E2%80%99%3D2%2C8%C2%B710-4+M+en+%5BNH3%5D%3D10+M.jpg "Efecto de la estabilidad del complejo formado. Cuanto más perfecto sea el complejo formado, tanto más fácilmente se disolverá el precipitado. AgI (Ps=10-16,1): S’=2,8·10-4 M en [NH3]=10 M. Se recurre a un complejo más perfecto (Kd menor) Para [CN-]=10 M:")
136
C) Proceso de oxidación-reducción
Solubilización de precipitados C) Proceso de oxidación-reducción El CuS no se disuelve en H2SO4 o en HCl diluidos, debido a su bajo Ps. Pero se disuelve en HNO3 gracias al poder oxidante de este ácido. Es frecuente solubilizar los sulfuros por una acción conjunta de cambio en el número de oxidación y la formación de un complejo.
 Proceso de oxidación-reducción. El CuS no se disuelve en H2SO4 o en HCl diluidos, debido a su bajo Ps. Pero se disuelve en HNO3 gracias al poder oxidante de este ácido. Es frecuente solubilizar los sulfuros por una acción conjunta de cambio en el número de oxidación y la formación de un complejo.")
137
D) Formación de compuestos más insolubles
Solubilización de precipitados D) Formación de compuestos más insolubles Este fenómeno se utiliza para liberar el anión de sales de Ag difícilmente solubles, para su posterior identificación (p. ej.: Cl-, Br-, I- y SCN-). También se puede conseguir una extensiva transferencia de un compuesto a otro más soluble, si se adiciona cantidad suficiente de reactivo.
 Formación de compuestos más insolubles. Este fenómeno se utiliza para liberar el anión de sales de Ag difícilmente solubles, para su posterior identificación (p. ej.: Cl-, Br-, I- y SCN-). También se puede conseguir una extensiva transferencia de un compuesto a otro más soluble, si se adiciona cantidad suficiente de reactivo.")
138
BaSO4 arrastra siempre CaSO4
Precipitaciones inducidas La precipitación inducida consiste en el arrastre de precipitados, o la aparición insólita de los mismos, en medios que -teóricamente- son disolventes de los mismos. 1) Co-precipitación: la impurificación del precipitado tiene lugar durante su formación. BaSO4 arrastra siempre CaSO4 2) Post-precipitación: el precipitado sufre una impurificación con posterioridad a su formación. El Zn2+ es capaz de precipitar ZnS sobre el HgS
 Co-precipitación: la impurificación del precipitado tiene lugar durante su formación. BaSO4 arrastra siempre CaSO4. 2) Post-precipitación: el precipitado sufre una impurificación con posterioridad a su formación. El Zn2+ es capaz de precipitar ZnS sobre el HgS.")
139
Adsorción Oclusión Compuesto químico Coprecipitación Precipitaciones
Precipitaciones inducidas Precipitaciones inducidas Adsorción Oclusión Compuesto químico Coprecipitación Postprecipitación

140
Equilibrios de oxidación-reducción.
Tema 4. Equilibrios de oxidación-reducción Equilibrios de oxidación-reducción. Reacciones espontáneas y forzadas: celdas electrolíticas. Electrodos de 1ª y 2ª especie: electrodos de referencia. Constantes de equilibrio y potenciales estándar. Dismutación. Oxidantes y reductores de uso más frecuente.

141
Reacción red-ox: transferencia o intercambio de electrones.
Conceptos generales Reacción red-ox: transferencia o intercambio de electrones. Una sustancia (el reductor) los cede, y otra sustancia (el oxidante) los acepta. Oxidación: pérdida de uno o más electrones. Reducción: ganancia de electrones. Ox1 + e- Red1 (semirreacción) Red2 e- + Ox2 (semirreacción) Ox1 + Red2 Red1 + Ox2
 los cede, y otra sustancia (el oxidante) los acepta. Oxidación: pérdida de uno o más electrones. Reducción: ganancia de electrones. Ox1 + e- Red1 (semirreacción) Red2 e- + Ox2 (semirreacción) Ox1 + Red2 Red1 + Ox2.")
142
CELDAS ELECTROQUÍMICAS
Galvánicas (voltaicas, pilas) Tiene lugar una reacción química, de forma espontánea, produciendo corriente eléctrica. CELDAS ELECTROQUÍMICAS Electrolíticas El paso de una corriente eléctrica fuerza una reacción química. En ambos tipos de celdas, el electrodo donde tiene lugar la oxidación se denomina ÁNODO, y en el que ocurre la reducción es el CÁTODO.
 Tiene lugar una reacción química, de forma espontánea, produciendo corriente eléctrica. CELDAS. ELECTROQUÍMICAS. Electrolíticas. El paso de una corriente eléctrica fuerza una reacción química. En ambos tipos de celdas, el electrodo donde tiene lugar la oxidación se denomina ÁNODO, y en el que ocurre la reducción es el CÁTODO.")
143
ánodo / disolución / cátodo
Celdas galvánicas A Fe2+ + Ce4+ Fe3+ + Ce3+ e- Fe3++ e- + e- Ce3+ KCl sat. Cl- K+ Fe2+ Ce4+ CÁTODO (+) ÁNODO (-) ánodo / disolución / cátodo
 ÁNODO (-) ánodo / disolución / cátodo.")
144
Celdas galvánicas

145
Galvánicas Electrolíticas ÁNODO: NEGATIVO (donde se generan los e-)
Celdas y convenios Galvánicas ÁNODO: NEGATIVO (donde se generan los e-) CÁTODO: POSITIVO (hacia donde migran los e-) Electrolíticas ÁNODO: POSITIVO (hacia donde migran los aniones) CÁTODO: NEGATIVO (hacia donde migran los cationes)
 CÁTODO: POSITIVO (hacia donde migran los e-) Electrolíticas. ÁNODO: POSITIVO (hacia donde migran los aniones) CÁTODO: NEGATIVO (hacia donde migran los cationes)")
146
Electrodo Normal de Hidrógeno (E.N.H.)
2 H e- H2 (H e 1/2 H2) Todos los potenciales estándar de los distintos semisistemas están medidos frente a este electrodo (ánodo).
 Todos los potenciales estándar de los distintos semisistemas están medidos frente a este electrodo (ánodo).")
147
Para el caso general: a Ox + n e- b Red
Ecuación de Nernst En 1889 Nernst formuló una expresión que relaciona el potencial de una semicelda con las concentraciones de las especies que contiene. Para el caso general: a Ox + n e- b Red Para T= 298 K:

148
Potencial Estándar y Potencial Formal
El potencial estándar, E0, es el potencial del electrodo cuando todos los reactivos y productos que aparecen en una semi-reacción tienen actividad unidad. El potencial formal, E0’, es el potencial estándar para unas condiciones experimentales determinadas y definidas.

149
Electrodos de referencia
E = f([Cl-]) Ag/AgClsat., KCl//
 Ag/AgClsat., KCl//")
150
Electrodos de segunda especie

151
Hg/Hg2Cl2sat., KCl// E = f([Cl-])
Electrodo de referencia de calomelanos Hg/Hg2Cl2sat., KCl// E = f([Cl-])
![Hg/Hg2Cl2sat., KCl// E = f([Cl-])](http://slideplayer.es/slide/3290951/11/images/151/Hg%2FHg2Cl2sat.%2C+KCl%2F%2F+E+%3D+f%28%5BCl-%5D%29.jpg "Electrodo de referencia de calomelanos. Hg/Hg2Cl2sat., KCl// E = f([Cl-])")
152
Para una reacción completa:
Ecelda = Ecátodo - Eánodo Ecelda=

153
Ecelda= - Ecelda= - - = K Ecelda = Ecátodo - Eánodo
Para una reacción completa: Ecelda = Ecátodo - Eánodo Ecelda= Ecelda= = K En el equilibrio: Ecelda = 0

154
Ej.: calcule la constante de equilibrio para la reacción
En el equilibrio: E1 = E2

155
Ej.: calcule la constante de equilibrio para la reacción
En el equilibrio: E1 = E2 Disponer de una tabla de potenciales normales es tener una tabla de K

156
E0’ pH Ej.: As(V)/As(III) E0’ I3-/3I- AsO43-/AsO33-
Efecto del pH sobre el potencial A) Tanto los iones H+ como OH- intervienen directamente en múltiples semirreacciones, cuyo potencial puede verse afectado por las variaciones del pH de la disolución. Ej.: As(V)/As(III) E0’ E0’ pH I3-/3I- AsO43-/AsO33-
 Tanto los iones H+ como OH- intervienen directamente en múltiples semirreacciones, cuyo potencial puede verse afectado por las variaciones del pH de la disolución. Ej.: As(V)/As(III) E0’ E0’ pH. I3-/3I- AsO43-/AsO33-")
157
Reacción global: Ox + ne- + H+ HRed+
Efecto del pH sobre el potencial B) Alguna de las especies del par redox puede sufrir una reacción secundaria de protonación, lo que influye indirectamente en el valor del potencial redox. Ox + n e- Red Red + H+ HRed+ Reacción global: Ox + ne- + H+ HRed+ (o bien: Ox + ne- + H2O HRed+ + OH-) Que ahora podemos sustituir en la ecuación de Nernst:
 Alguna de las especies del par redox puede sufrir una reacción secundaria de protonación, lo que influye indirectamente en el valor del potencial redox. Ox + n e- Red. Red + H+ HRed+ Reacción global: Ox + ne- + H+ HRed+ (o bien: Ox + ne- + H2O HRed+ + OH-) Que ahora podemos sustituir en la ecuación de Nernst:")
158
E0’ Situaciones extremas:
Efecto del pH sobre el potencial E0’ Situaciones extremas: a) [H+]<<Ka; pH>>pKa; [H+]/Ka <<1 E0’ = E0; el pH no modifica el potencial (predominio de Red) b) [H+]>>Ka; pH<<pKa; [H+]/Ka>>1 El potencial aumenta con [H+] pH pKa E0’ Red HRed
 [H+]<<Ka; pH>>pKa; [H+]/Ka <<1. E0’ = E0; el pH no modifica el potencial (predominio de Red) b) [H+]>>Ka; pH<<pKa; [H+]/Ka>>1. El potencial aumenta con [H+] pH. pKa. E0’ Red. HRed.")
159
E0’ S + 2 e- S2- E = E0 - 0,03 log [S2-]
Ejemplo: Estudio del sistema S2-/S S + 2 e- S2- E = E ,03 log [S2-] Al aumentar [H+], el sistema se volverá más oxidante. E0’
![E0’ S + 2 e- S2- E = E0 - 0,03 log [S2-]](http://slideplayer.es/slide/3290951/11/images/159/E0%E2%80%99+S+%2B+2+e-+%EF%81%84+S2-+E+%3D+E0+-+0%2C03+log+%5BS2-%5D.jpg "Ejemplo: Estudio del sistema S2-/S. S + 2 e- S2- E = E0 - 0,03 log [S2-] Al aumentar [H+], el sistema se volverá más oxidante. E0’")
160
Ejemplo: cálculo del E en una disolución de [S2-]T= 1 M
a) Si [H+]<<K2; pH>>pK2: E0’ = E0 y la especie predominante es el S2-. b) Si 10-7>[H+]>10-13; 7<pH<13: E0’ = E0 + 0,03 log ([H+]/K2) = E0 - 0,03 log K2 - 0,03 pH, y la especie predominante es el HS-.
![Ejemplo: cálculo del E en una disolución de [S2-]T= 1 M](http://slideplayer.es/slide/3290951/11/images/160/Ejemplo%3A+c%C3%A1lculo+del+E+en+una+disoluci%C3%B3n+de+%5BS2-%5DT%3D+1+M.jpg "a) Si [H+]<<K2; pH>>pK2: E0’ = E0 y la especie predominante es el S2-. b) Si 10-7>[H+]>10-13; 7<pH<13: E0’ = E0 + 0,03 log ([H+]/K2) = E0 - 0,03 log K2 - 0,03 pH, y la especie predominante es el HS-.")
161
c) Si [H+]>K1; pH<7:
E0’ = E0 - 0,03 log K1K2 - 0,06 pH, siendo la especie predominante el H2S.
![c) Si [H+]>K1; pH<7:](http://slideplayer.es/slide/3290951/11/images/161/c%29+Si+%5BH%2B%5D%3EK1%3B+pH%3C7%3A.jpg "E0’ = E0 - 0,03 log K1K2 - 0,06 pH, siendo la especie predominante el H2S.")
162
Efecto del pH sobre el potencial
En ocasiones el pH influye porque modifica el potencial por precipitación de alguna de las formas del par redox. pH E Fe3+ Fe(OH)3 Fe(OH)2 8 2,5
3. Fe(OH) ,5.")
163
Efecto de las reacciones de complejación
La presencia de complejantes en el medio de reacción hace variar el potencial de una semirreacción como consecuencia de la disminución de la concentración de una o varias de las especies iónicas.

164
2 Cu+ Cu0 + Cu2+ Cu+ + e- Cu0; E0 = 0,521 V
Dismutación El fenómeno de la dismutación, desproporcionamiento o auto oxidación-reducción, consiste en la autotransformación de un ión en un estado de oxidación intermedio para originar, a sus expensas, dos estados de oxidación: uno más oxidado y otro más reducido. Cu0 Cu2+ Cu+ HNO3 NO HNO2 Cl- ClO3- ClO- Condición de dismutación: Cu+ + e- Cu0; E0 = 0,521 V Cu2+ + e- Cu+; E0 = 0,153 V 2 Cu+ Cu0 + Cu2+ E =0,521-0,153=0,368 V

165
Oxidantes y reductores más utilizados

166
Extracciones sucesivas. Rendimiento de la extracción.
Tema 5. Equilibrios de reparto y de intercambio iónico Ley de reparto. Extracciones sucesivas. Rendimiento de la extracción. Naturaleza de los cambiadores iónicos. Equilibrio de intercambio y factores que le afectan.

167
Ley de Reparto Un soluto A se reparte entre dos fases inmiscibles entre sí ( fase acuosa y orgánica), en función de su solubilidad en ambas fases: La proporción de actividades, o concentraciones, del soluto en ambas fases es una constante que recibe el nombre de coeficiente de distribución o de reparto.

168
La aplicación de la ley de distribución exige que se haya establecido el equilibrio dinámico entre las fases: el tiempo de equilibración es un factor importante. La transferencia del soluto depende de : la superficie de la interfase la velocidad de difusión del soluto hasta homogeneizar su concentración Ambas se ven favorecidas por la agitación. KD es constante (para cada T y ), pero la extensión de la extracción depende de : valor relativo de los volúmenes de las fases inmiscibles número de veces que se realiza la extracción con porciones frescas de fase extractiva.
, pero la extensión de la extracción depende de : valor relativo de los volúmenes de las fases inmiscibles. número de veces que se realiza la extracción con porciones frescas de fase extractiva.")
169
Vo Va C: concentración inicial de soluto en fase acuosa
Va: volumen de fase acuosa Vo: volumen de fase orgánica En el equilibrio: C1: concentración de soluto en fase acuosa C1’: concentración de soluto en fase orgánica Va Vo

170
Balance de materia: KD sea grande r sea elevado
Volúmenes relativos de las fases Balance de materia: Dividiendo numerador y denominador por Va: La extracción se favorecerá (C1 pequeña) cuando: KD sea grande r sea elevado
 cuando: KD sea grande. r sea elevado.")
171
C: concentración inicial de soluto en fase acuosa
Extracciones sucesivas Extracciones sucesivas de soluto presente en un volumen Va de fase acuosa con n porciones de volumen Vo de fase orgánica. C: concentración inicial de soluto en fase acuosa C1, C2, C3,......, Cn: concentraciones en la fase acuosa después de 1,2,3,..., n extracciones. C1’, C2’, C3’,...,Cn’: concentraciones en la fase orgánica tras las extracciones sucesivas. Después de la primera extracción sabemos que se cumple: Cuando se ha separado la primera fase orgánica (con una concentración C1’), y se equilibra con una nueva porción de Vo fresco:
, y se equilibra con una nueva porción de Vo fresco:")
172
c a d b a) I2; b) n=1; Vo=40 mL; c y d) n=2; Vo = 20 mL ac. org.
Extracción de I2 co HCCl3 c a d b ac. org. a) I2; b) n=1; Vo=40 mL; c y d) n=2; Vo = 20 mL
 I2; b) n=1; Vo=40 mL; c y d) n=2; Vo = 20 mL.")
173
c a d b a) I2; b) n=1; Vo=40 mL; c y d) n=2; Vo = 20 mL ac. org.
Extracción de I2 co HCCl3 c a d b ac. org. a) I2; b) n=1; Vo=40 mL; c y d) n=2; Vo = 20 mL
 I2; b) n=1; Vo=40 mL; c y d) n=2; Vo = 20 mL.")
174
c a d b a) I2; b) n=1; Vo=40 mL; c) n=2; Vo = 20 mL; ac. org.
Extracción de I2 co HCCl3 c a d b ac. org. a) I2; b) n=1; Vo=40 mL; c) n=2; Vo = 20 mL;
 I2; b) n=1; Vo=40 mL; c) n=2; Vo = 20 mL;")
175
Extracción de I2 co HCCl3 c a d b ac. org. a) I2; b) n=1; Vo=40 mL; c) n=2; Vo = 20 mL; d) n=4; Vo = 10 mL
 I2; b) n=1; Vo=40 mL; c) n=2; Vo = 20 mL; d) n=4; Vo = 10 mL.")
176
Extracción de I2 co HCCl3 c a d b ac. org. d b c a ac. a) I2; b) n=1; Vo=40 mL; c) n=2; Vo = 20 mL; d) n=4; Vo = 10 mL Fases acuosas: a) I2; b) n=1; Vo=40 mL; c) n=2; Vo = 20 mL; d) n=4; Vo = 10 mL
 I2; b) n=1; Vo=40 mL; c) n=2; Vo = 20 mL; d) n=4; Vo = 10 mL.")
177
Si en a) el Vo fuese de 50 mL: r= 0,5 C1 = 0,033 M; R = 83,5%
Ejemplo Se extraen 100 mL de una disolución 0,2 M de un compuesto AB a) con 100 mL de cloroformo; b) con 2 porciones de 50 mL de cloroformo; c) con 5 porciones de 20 mL de cloroformo. Calcule el factor de recuperación en cada caso. DATO: KD AB = 10 Si en a) el Vo fuese de 50 mL: r= 0,5 C1 = 0,033 M; R = 83,5%
 con 100 mL de cloroformo; b) con 2 porciones de 50 mL de cloroformo; c) con 5 porciones de 20 mL de cloroformo. Calcule el factor de recuperación en cada caso. DATO: KD AB = 10. Si en a) el Vo fuese de 50 mL: r= 0,5 C1 = 0,033 M; R = 83,5%")
178
Rendimiento de la extracción
Fracción (tanto por uno) o porcentaje (tanto por ciento) de la cantidad total de soluto extraída en la fase orgánica, Q, en relación con la cantidad total de soluto puesta en juego, Q’, que está inicialmente en la fase acuosa. n
 o porcentaje (tanto por ciento) de la cantidad total de soluto extraída en la fase orgánica, Q, en relación con la cantidad total de soluto puesta en juego, Q’, que está inicialmente en la fase acuosa. n.")
179
Cuando se realiza una sola operación de extracción, y los volúmenes de ambas fases son iguales, n= r=1: 1) KD < 1 R < 50%; ) KD > 1 R > 50% 3) R = 99,9% KD 103; 4) R = 0,1% KD 10-3
 KD < 1 R < 50%; 2) KD > 1 R > 50% 3) R = 99,9% KD 103; 4) R = 0,1% KD")
180
Relación de Distribución
Para que se cumpla la ley de distribución, es preciso que el soluto se encuentre en la misma forma química en ambas fases. Para las situaciones donde esta condición no se cumple, por estar el soluto formando especies moleculares, iónicas, complejos, etc., se define la relación de distribución o coeficiente de reparto condicional.

181
Mn+ + n (HR)o (MRn)o + n H+
Influencia del pH en la extracción Fase acuosa Fase orgánica HR MRn KD(HR) KD(MRn) HR R- + H+ Ka nR- + M n MRn n Mn+ + n (HR)o (MRn)o + n H+
 KD(MRn) HR R- + H+ Ka. nR- + M n+ MRn. n. Mn+ + n (HR)o (MRn)o + n H+")
182
1) Equilibrio de reparto del reactivo
2) Formación del quelato 3) Reparto del quelato entre las fases
 Formación del quelato. 3) Reparto del quelato entre las fases.")
183
1) Equilibrio de reparto del reactivo
2) Formación del quelato 3) Reparto del quelato entre las fases 1. Suponiendo que no hay reacciones secundarias del reactivo en fase orgánica: [HR]o=[HR]o 2. Si en fase acuosa solo tiene lugar la disociación del reactivo: [HR]a= [HR]a + [R-]a El reparto del reactivo vendrá regulado exclusivamente por el pH Una extracción efectiva requiere que pH > pKa
 Formación del quelato. 3) Reparto del quelato entre las fases. 1. Suponiendo que no hay reacciones secundarias del reactivo en fase orgánica: [HR]o=[HR]o. 2. Si en fase acuosa solo tiene lugar la disociación del reactivo: [HR]a= [HR]a + [R-]a. El reparto del reactivo vendrá regulado exclusivamente por el pH. Una extracción efectiva requiere que pH > pKa.")
184
3) Reparto del quelato entre las fases
 Reparto del quelato entre las fases")
185
Mn+ + n R- MRn MRn Mn+ + n R- MRn n KD MRn Kext
2) Formación del quelato Mn+ + n R MRn MRn n KD MRn Mn+ + n R MRn Kext
 Formación del quelato. Mn+ + n R- MRn. MRn. n. KD MRn. Mn+ + n R- MRn. Kext.")
186
1. Separación de componentes de una disolución
Aplicaciones analíticas 1. Separación de componentes de una disolución eliminación de interferentes preconcentración del analito 2. Obtención de patrones de alto grado de pureza

187
Intercambio de cantidades equivalentes de iones.
Intercambio Iónico 1854: Way y Thomson observaron que cierto tipo de suelos podían cambiar los iones Ca2+ por iones NH4+, cuando se hacía pasar (NH4)2SO4 a través de una columna de tierra. Intercambio de cantidades equivalentes de iones. Ciertos iones eran mucho más intercambiables que otros. Los responsables del cambio eran aluminosilicatos, que actuaban de soporte físico. La T no afectaba apreciablemente al proceso.
2SO4 a través de una columna de tierra. Intercambio de cantidades equivalentes de iones. Ciertos iones eran mucho más intercambiables que otros. Los responsables del cambio eran aluminosilicatos, que actuaban de soporte físico. La T no afectaba apreciablemente al proceso.")
188
H+ SO3- + Na+Cl- Na+SO3- + H+Cl-
Cambiador iónico “sólido poroso que tiene la propiedad de fijar los iones contenidos en una disolución que baña sus poros, intercambiándolos por iones del mismo signo que pertenecen a la estructura del sólido cambiador” H+ SO3- + Na+Cl- Na+SO H+Cl- H+ , Na+ : contraiones SO3-: grupo funcional Cl-: co-ión

189
Estructura hidrofílica de forma regular y reproducible
Propiedades del Cambiador Ideal Estructura hidrofílica de forma regular y reproducible Tamaño de partícula uniforme Rápida velocidad de intercambio Químicamente estable Físicamente estable (resistencia mecánica al desgaste) Térmicamente estable
 Térmicamente estable.")
190
aluminosilicatos: permutitas óxidos hidratados: ZrO2
Cambiadores Inorgánicos Cambiadores Orgánicos (resinas) aluminosilicatos: permutitas óxidos hidratados: ZrO2 sales ácidas de elementos polivalentes sales de heteropoliácidos sólidos sintéticos: apatitas, ferrocianuros
 aluminosilicatos: permutitas. óxidos hidratados: ZrO2. sales ácidas de elementos polivalentes. sales de heteropoliácidos. sólidos sintéticos: apatitas, ferrocianuros.")
191
Cationes, aniones y sistema periódico de los elementos.
Tema 6. Propiedades periódicas de los elementos Cationes, aniones y sistema periódico de los elementos. Estabilidad de cationes, aniones y oxoaniones. El color en Química Analítica: grupos cromóforos y auxocromos. Sensibilidad y selectividad de las reacciones analíticas.

192
Cationes y sistema periódico
Formación de cationes electronegatividad

193
Elementos no formadores de cationes
Elementos formadores de aniones sencillos

194
Estabilidad de los cationes
La fuerza con la que un catión retiene las moléculas del agua solvatada depende de: tamaño del ión valor de la carga positiva Carga positiva/radio; q/r q/r =58,8 Los cationes son siempre ácidos o neutros Estabilidad de cationes sencillos q/r acidez

195
Cationes del tercer período

196
Be, Al, Ti(IV), Cr(III), Zn Sn(II), Sn(IV) Sb(III), Sb(V)
Elementos formadores de hidróxidos anfóteros Be, Al, Ti(IV), Cr(III), Zn Sn(II), Sn(IV) Sb(III), Sb(V) Pb(II), Pb(IV)
, Cr(III), Zn. Sn(II), Sn(IV) Sb(III), Sb(V) Pb(II), Pb(IV)")
197
Cationes de los alcalinotérreos
q/r Estabilidad

198
Valores próximos de q/r implican un comportamiento analítico semejante en disolución acuosa:
Be2+ (57,2) y Al3+(58,8) Cuando un elemento puede actuar con distintos grados de oxidación, los cationes que origina son tanto más estables en disolución acuosa cuanto menor es el grado de oxidación del elemento: Cr2+, Cr3+, Cr6+
 y Al3+(58,8) Cuando un elemento puede actuar con distintos grados de oxidación, los cationes que origina son tanto más estables en disolución acuosa cuanto menor es el grado de oxidación del elemento: Cr2+, Cr3+, Cr6+")
199
Estado de oxidación del Manganeso

200
S2- (r=0,184 nm): S2- + H2O HS- + OH- HS- + H2O H2S + OH-
Aniones elementales Debido a su volumen, sólo los aniones muy cargados y no excesivamente grandes ejercerán su acción atractiva sobre los dipolos del agua. S2- (r=0,184 nm): S2- + H2O HS- + OH- HS- + H2O H2S + OH- Anión básico Cl- (r=0,181 nm): estable a todo pH Anión neutro Los aniones son siempre básicos o neutros
: S2- + H2O HS- + OH- HS- + H2O H2S + OH- Anión básico. Cl- (r=0,181 nm): estable a todo pH. Anión neutro. Los aniones son siempre básicos o neutros.")
201
Estabilidad aniones sencillos
Aumenta q/r Basicidad aniones

202
Un elemento formará oxoaniones más estables cuanto más fuertemente retenga al O2-(base), o sea, cuanto mayor sea su q/r (mayor carácter ácido posea). Estabilidad oxoaniones Cuando un elemento pueda formar diversos oxoaniones, serán más estables los correspondientes a un mayor grado de oxidación: ClO-, ClO2-, ClO3-, ClO4- Para un elemento determinado, los cationes serán tanto más estables cuanto menor sea su estado de oxidación, en tanto que la estabilidad de sus aniones se incrementa con el grado de oxidación.

203
El color es la energía electromagnética comprendida entre 400 y 700nm.
El Color en Química Analítica “En el fenómeno cromático se superponen componentes físicos, fisiológicos y psíquicos”. El color es la energía electromagnética comprendida entre 400 y 700nm.

208
El espectro visible

209
Color de los iones sencillos

210
Color de los iones sencillos

211
Color de los iones sencillos
Forman cationes elementales coloreados aquellos átomos que al perder sus electrones de valencia para originar el catión, les queda -en la envolvente más externa del ión- orbitales del tipo d incompletos, es decir, con menos de 10 electrones, mientras que si dichos orbitales d llegan a la saturación electrónica (10 electrones), o no existen como capa externa, los iones formados (aniones o cationes) son siempre incoloros.
, o no existen como capa externa, los iones formados (aniones o cationes) son siempre incoloros.")
212
Cr3+ en medio HCl Cr3+ en medio HNO3

213
Cromóforos Witt (1876): ciertos grupos funcionales no saturados proporcionan color a la estructura carbonada en que se encuentran: cromóforos (“portador de color”).
: ciertos grupos funcionales no saturados proporcionan color a la estructura carbonada en que se encuentran: cromóforos ( portador de color ).")
214
Auxocromos “Radicales que por sí mismos no dan color a una molécula, pero pueden exaltar la acción de un cromóforo”.

215
Reactivo específico del Zr4+
Preparación de reactivos específicos Ti4+ Zr4+ Sn4+ Th4+ Precipitados blancos Precipitado pardo Reactivo específico del Zr4+

216
Precipitados blanco-amarillentos
Preparación de reactivos específicos Ag+ Hg2+ Au3+ Precipitados blanco-amarillentos

217
Sensibilidad de una reacción analítica
Expresa la cantidad o concentración mínima que se puede identificar con una determinada reacción analítica. Límite de identificación: cantidad mínima de sustancia que puede reconocerse en un ensayo. Concentración límite: mínima concentración de especie a la que un ensayo resulta positivo, D. Dilución límite: dilución por encima de la cual un ensayo deja de ser positivo. Es el valor inverso de la concentración límite: 1/D

218
Ejemplo La reacción de identificación del Cu(II) con cuprón (-benzoinoxima, H5C6-CH(OH)-C(=NOH)-C6H5) posee un límite de identificación de 0,12 g cuando el ensayo se lleva a cabo en papel de filtro (0,03 mL). Calcule la sensibilidad expresada como D, pD, ppm y %. Indíquese también el límite de dilución.
 con cuprón (-benzoinoxima, H5C6-CH(OH)-C(=NOH)-C6H5) posee un límite de identificación de 0,12 g cuando el ensayo se lleva a cabo en papel de filtro (0,03 mL). Calcule la sensibilidad expresada como D, pD, ppm y %. Indíquese también el límite de dilución.")
219
Clasificación de las reacciones
No siempre las reacciones más sensibles son las mejores: búsqueda de compromiso entre la sensibilidad y el fin analítico perseguido.

220
1. Desplazamiento de la reacción: A + R P
Factores que afectan a la sensibilidad 1. Desplazamiento de la reacción: A + R P Fe3+ + SCN- FeSCN2+ rojo, muy poco estable Hg SCN- Hg(SCN)42- incoloro, muy estable 2. Influencia del color 3. Influencia de la técnica empleada Papel de filtro > tubo de ensayo > placa de gotas 4. Influencia de la extracción. Rosa pálido Azul en H3CCl 4.a) Desplazamiento del equilibrio 4.b) Concentración de la especie: Vo < Va 4.c) Cambio y aumento del color 4.d) Separación 5. Influencia de sustancias extrañas Interferentes o consumidores de reactivo. Fe3+ con SCN-. Hg2+ consume reactivo.
42- incoloro, muy estable. 2. Influencia del color. 3. Influencia de la técnica empleada. Papel de filtro > tubo de ensayo > placa de gotas. 4. Influencia de la extracción. Rosa pálido. Azul en H3CCl. 4.a) Desplazamiento del equilibrio. 4.b) Concentración de la especie: Vo < Va. 4.c) Cambio y aumento del color. 4.d) Separación. 5. Influencia de sustancias extrañas. Interferentes o consumidores de reactivo. Fe3+ con SCN-. Hg2+ consume reactivo.")
221
Exaltación de la reactividad
Fenómeno por el cual un reactivo que normalmente no actúa sobre un determinado analito, pasa a hacerlo en determinadas condiciones experimentales. Azul de molibdeno

222
* Modificación del poder oxidante o reductor
Exaltación de la reactividad * Modificación del poder oxidante o reductor Fe2+: aumenta su poder reductor en presencia de complejantes del Fe3+ (fluoruros, tartratos, AEDT,…). * Reacciones catalíticas Mejora en la cinética de la reacción. El catalizador no experimenta un cambio permanente. Catalizada por la Ag+
. * Reacciones catalíticas. Mejora en la cinética de la reacción. El catalizador no experimenta un cambio permanente. Catalizada por la Ag+")
223
* Reacciones inducidas
Exaltación de la reactividad * Reacciones inducidas El inductor experimenta un cambio permanente. Reacción inductora 2 SO O2 2 SO42- Reacción inducida 4 Ni(OH)2 + O H2O 4 Ni(OH)3 Actor: O2 Inductor: SO32- Aceptor: Ni(OH)2
2 + O2 + 2 H2O 4 Ni(OH)3. Actor: O2. Inductor: SO32- Aceptor: Ni(OH)2.")
224
* Reacciones sensibilizadas
Exaltación de la reactividad * Reacciones sensibilizadas Espectrofotometría de absorción: A · C M + n L MLn ; puede ser baja generalmente aumenta; complejos más estables y más fácilmente extraíbles. M + n L + m P MLnPm M + N + n L MNLn Ejemplo: H3PMo12O40, amarillo, = 600 + V(V), amarillo naranja, = 2.500
, amarillo naranja, =")
225
Selectividad de las reacciones
* Grupos selectivos Cu(II), Mo (VI)
, Mo (VI)")
226
* Modificación de la porción no selectiva
Selectividad de las reacciones * Modificación de la porción no selectiva 3 No reacciona con el Fe2+

227
* Modificación de la solubilidad
Selectividad de las reacciones * Modificación de la solubilidad Soluble en agua. Insoluble en disolventes no polares

Presentaciones similares





3 + Ba++ BaSO4 analito (Fe3+) no forma parte del producto>")



.>")









