Descargar la presentación
La descarga está en progreso. Por favor, espere

1
BASES MOLECULARES DE LAS ENFERMEDADES
GENÉTICAS Dr. José Antonio Nastasi Catanese

2
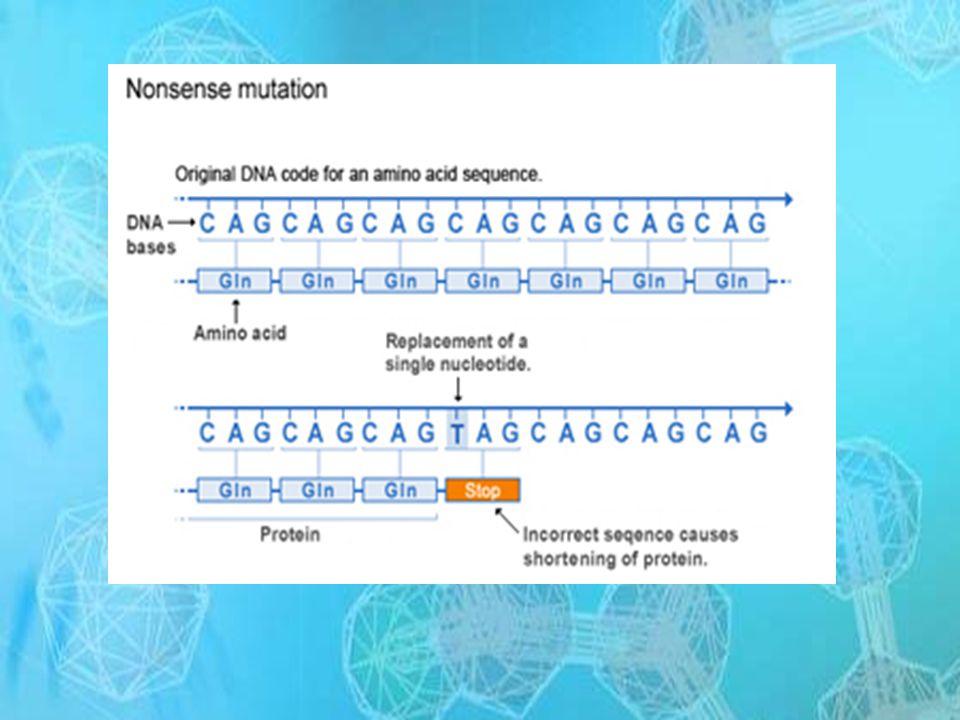
MUTACIÓN Se define como cualquier cambio que ocurre en la molécula de ADN

3
TIPOS DE MUTUACIONES PUNTUALES

4
SUSTITUCIÓN

5
DELECIÓN, PERDIDA O ELIMINACIÓN
CORRIMIENTO DEL MARCO DE LECTURA

6
INSERCIÓN CORRIMIENTO DEL MARCO DE LECTURA

8
Una mutación puede alterar o no el código genético que codifica para una proteína determinada.
Si la alteración del código determina una modificación de la estructura primaria de la proteína puede haber una modificación de la función de esa proteína, aumentando, disminuyendo o anulando dicha función Si la función de la proteína dada está alterada, las manifestaciones clínicas dependerán de la función de dicha proteína.

9
En el caso de proteínas estructurales o que se combinan con otras cadenas polipeptídicas para dar origen a un complejo funcional, la anomalía de uno solo de los dos genes normales puede dar origen a una enfermedad (Dominante). Si el gene alterado codifica para una enzima, será necesario la afectación de los dos genes (Recesiva)
")
10
FUNCIONES DE LAS PROTEINAS
Enzimas Transporte Estructura de células y órganos Homeostasia extracelular Control de crecimiento y diferenciación Metabolismo y comunicación intercelular

11
ERRORES CONGÉNITOS DEL METABOLISMO
ALCAPTONURIA

12
Acido homogentísico ALCAPTONURIA
Ocronosis en los ojos y pabellón auricular Acido homogentísico

13
DEFECTOS DE LAS PROTEINAS ESTRUCTURALES
INTRACELULARES:QUERATINA:EPIDERMOLISIS AMPOLLAR EXTRACELULARES:COLAGENO:OSTEOGENESIS IMPERFECTA EXTRACELULARES FORMADORAS DE RECEPTORES: HIPERCOLESTEROLEMIA FAMILIAR EXTRACELULARES FORMADORAS DE CANALES DE MEMBRANA: FIBROSIS QUISTICA EXTRACELULARESTRANSPORTADORAS:HEMOGLOBINOPATIAS

14
DEFECTOS EN EL TRANSPORTE A TRÁVES
DE LA MEMBRANA CITOPLASMATICA Fibrosis Quística Es una enfermedad autosómica recesiva, que afecta 1 de cada 2000 recién nacidos vivos en las poblaciones caucásicas Es una enfermedad generalizada que compromete las glándulas exocrinas y sudoríparas de todo el cuerpo, a causa de un defecto en el transporte del anión cloro en las membranas celulares de los epitelios glandulares

15
FIBROSIS QUISTICA MANIFESTACIONES RESPIRATORIAS La secreción mucosa del epitelio respiratorio es muy viscosa y obstruye las vias respiratorias , dificultando la función ventilatoria normal , provocando infecciones recurrentes , la destrucción del parénquima pulmonar, produciendo una enfermedad pulmonar obstructiva crónica y a su vez insuficiencia cardiaca

16
FIBROSIS QUISTICA La insuficiencia pancreática es debida a la secreción poco hidratada de los acinos que se estanca en los conductos, los dilata y produce quistes que por compresión producen la atrofia del tejido secretor , disminución de las enzimas pancreáticas produciendo desnutrición y anemia por mala absorción

17
Los niveles de los electrolitos
FIBROSIS QUISTICA Los niveles de los electrolitos Cl y Na en el sudor están elevados por encima de 60 mEq/L en el 100 % de los casos Ausencia del conducto deferente en la casi totalidad de los varones afectados

18
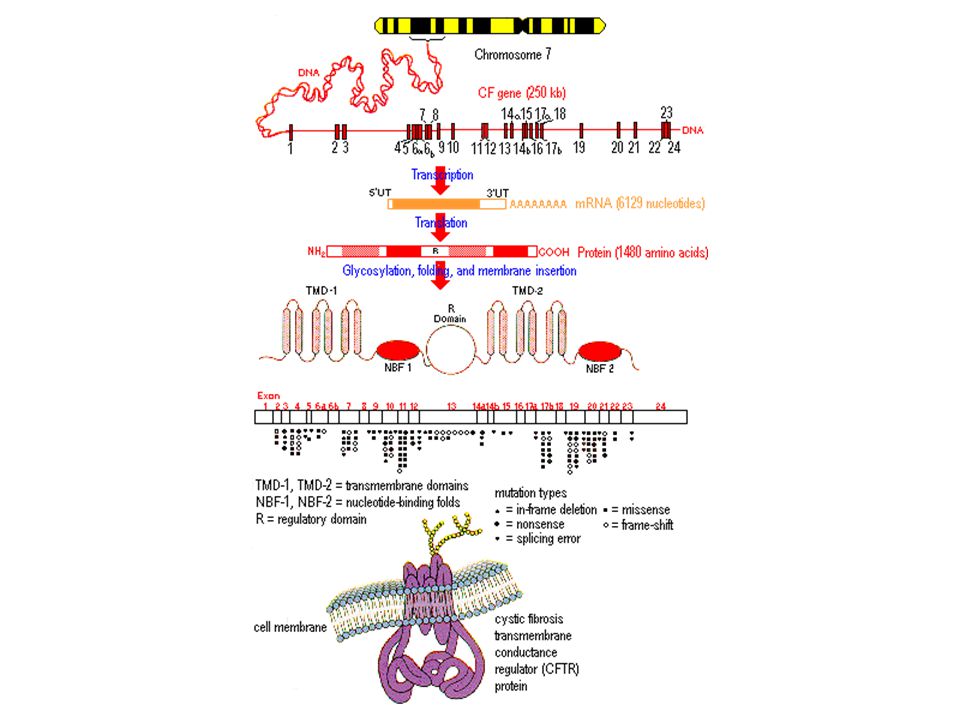
FIBROSIS QUISTICA El gen de la fibrosis quística esta localizado en el brazo largo del cromosoma 7

19
PROTEÍNA CFTR Este gen es muy grande y codifica para una proteína de aminoácidos que se denomina regulador transmembranoso de la conductancia de la fibrosis quística . Esta proteína es un canal de cloro de la membrana citoplasmática

20
FIBROSIS QUISTICA Esta proteína es compleja y tiene cinco dominios, dos ligadores de nucleótidos, que se asocian con dos moléculas de ATP, el dominio regulador que se asocia con la proteinkinasa y los dos dominios llamados transmembranosos que forman las paredes del canal insertados en la doble capa lipídica de la membrana, dejando un pasaje central para el pasaje del ión CL

21
FIBROSIS QUISTICA Se han descrito mas de 400 mutaciones en el gen de la FQ, existiendo una mayor concentración en los exones 10 y 11, que codifican para el primer dominio ligador del ATP y una de estas mutaciones denominada delta F508 esta presente en el 70% de los casos y consiste en una deleción de tres bases En el exón 10, que ocasiona la pérdida del aminoácido fenilalanina correspondiente al codón 508

22
PATOGENIA DE LA ENFERMEDAD PULMONAR
FIBROSIS QUISTICA Mutación del gen RTFQ Tipos I y II Proteína RTFQ anormal Falla en el plegamiento Retención de iones y agua Canal de Cl no funcional Secreción mucosa anormal Poco hidratada Acumulación de secreción No movilizada por movimiento ciliar Hipoventilación pulmonar Disminución de luz en conductos Inflamación mucosa Infección por Psuedomonas Enfermedad pulmonar crónica Bronquiectasias, enfisema

24
DEFECTOS EN LAS PROTEINAS TRASPORTADORAS
HEMOGLOBINOPATIAS:DREPANOCITOSIS La drepanocitosis es una enfermedad autosómica recesiva que afecta a 1 de cada 500 nacidos vivos en las poblaciones de África Occidental y a las de origen africano en todo el continente americano. Esta enfermedad cursa con una anemia hemolítica crónica, con crisis dolorosas en las extremidades, ictericia, fiebre, esplenomegalia y se debe a una mutación puntual en el gen de la beta globina, una de las cadenas polipeptídicas que conforman la molécula de hemoglobina

25
DREPANOCITOSIS Los eritrocitos se observan alargados en forma de hoz y la hemoglobina tiene una migración anormal en la electroforesis

26
ESTRUCTURA DE LA HEMOGLOBINA
Son proteínas tetraméricas compuestas por cuatro cadenas polipeptídicas, dos de tipo alfa y dos de tipo beta , cada globina alberga un grupo hemo con hierro, que transporta el oxigeno

27
GENES DE LAS GLOBINAS HUMANAS
Los dos tipos de cadenas son codificados por genes situados en loci separados, los genes que codifican las cadenas alfa están situados en el cromosoma 16 y los que codifican las cadenas beta en el cromosoma 11

28
La mutación responsable de la drepanocitosis se produce en el primer exón de la cadena de globina beta, sustituyendo una timina por adenina, lo que cambia la codificación del sexto aminoácido de la globina beta, sustituyendo el ácido glutámico por una valina neutra

29
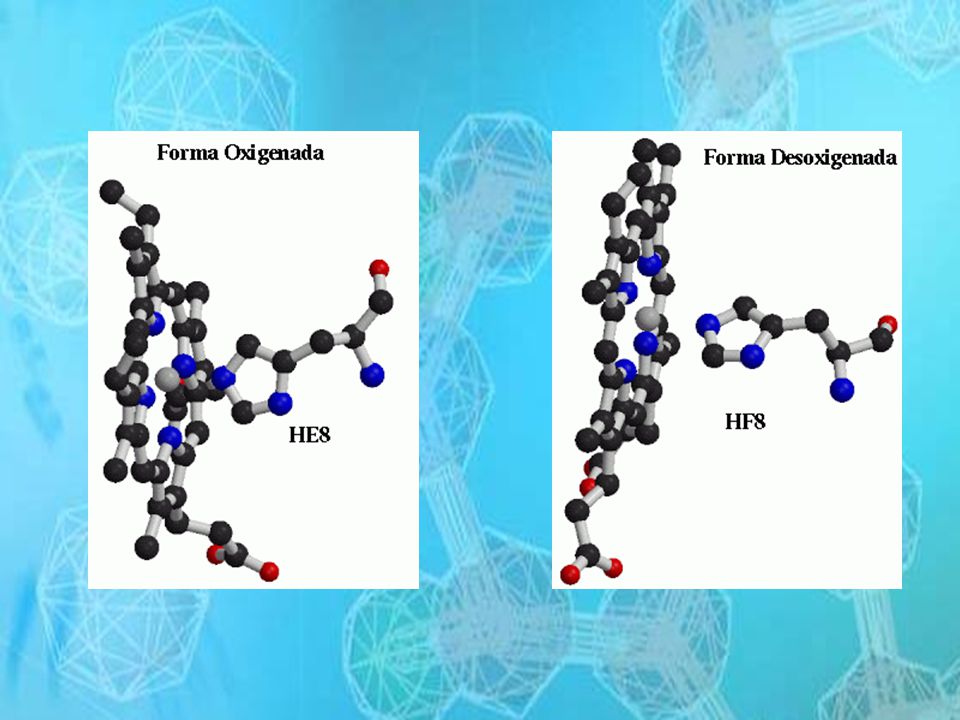
Las propiedades de la Hb dependen de la interacción de sus cuatro
unidades, las modificaciones en la estructura terciaria y cuaternaria pueden originar cambios en sus propiedades funcionales

30
Cuando disminuye la tensión de oxigeno la Hb S tiende a formar agregados moleculares en forma de columnas dentro del eritrocito provocando la forma alargada falciforme

32
Fisiopatología

33
TRASTORNOS QUE AFECTAN LAS PROTEINAS ESTRUCTURALES
DEFECTOS DEL COLAGENO:OSTEOGENESIS IMPERFECTA La familia de las proteínas del colágeno están constituidas por 19 tipos. Están compuestos por tres cadenas polipeptídicas(cadenas alfa) que pueden ser idénticas o diferentes, cada cadena tiene un gen diferente designado COL seguido del tipo de colágeno que determina
 que pueden ser idénticas o diferentes, cada cadena tiene un gen diferente designado COL seguido del tipo de colágeno que determina.")
34
Estructura del colágeno
Es una triple hélice muy regular de tres cadenas alfa, resultado de una secuencia repetida de tres aminoácidos, que comienzan por una glicina y otros dos aminoácidos X-Y, las tres cadenas se unen por el extremo carboxilo terminal

35
GEN COL1A1: CADENA ALFA 1 Situado en el cromosoma 17q 21.3 Se han encontrado mas de 80 mutaciones que afectan la codificación de la glicina inicial del dominio helicoidal central, la afección es mas severa cuando se acercan al dominio propéptido C

36
OSTEOGÉNESIS IMPERFECTA
Escleróticas azules Fracturas patológicas Las osteogénesis imperfectas son un grupo de trastornos hereditarios del colágeno tipo I que se caracterizan por fracturas múltiples, escleróticas azules, sordera y dentinogénesis imperfecta

37
OSTEOGÉNESIS IMPERFECTA
El tipo II presenta una fragilidad ósea extrema y una elevada mortalidad perinatal y fetal por fracturas intrauterinas

Presentaciones similares