Descargar la presentación
La descarga está en progreso. Por favor, espere

1
CAUSAS GENÉTICAS DEL COMPROMISO AXONAL
Dra. Susana Lara Dra. Alejandra Méndez Neuróloga Infantil Residente de Neurología Infantil ENERO 2014

2
Introducción El daño axonal es la base fisiopatológica de muchas enfermedades neurológicas degenerativas. El origen es complejo, ya que involucra múltiples vías moleculares y estructurales del axón. Su etiología puede ser genética o adquirida y su expresión fenotípica variable. La aproximación clínica y diagnóstica debe ser a través de la historia detallada, examen neurológico y estudio electrofisiologico.

3
Clasicamente el daño neuronal se divide en

4
Neurology® Clinical Practice 2011;76 (Suppl 2):S6–S13
Que? tipo de compromiso, sensitivo, motor o autonomico Donde? distal y simetrico, en EEII o solo superiores, compromete todo el cuerpo Cuando? evolucion temporal y progresion Neurology® Clinical Practice 2011;76 (Suppl 2):S6–S13
:S6–S13.")
5
Electrofisiología: Velocidad de conducción
Latencia VC > 38mm/seg Amplitud VC< 38 mm/seg La VC mide la fuerza y velocidad con que el impulso nervioso se propaga a traves del nervio periférico. En este este examen el potencial de acion se va a gatillar en un punto especifico con un estimulador bipolar, Los cambios se pueden ver en la VC 3 a 5 dias despues de l ainjuria. La velocidad es el tiempo que se demora en conducir el impulso desde que se provoca y esto se vera reflejado en la latencia En el daño axonal se observa un adisminucion en la amplitud. Inicialmente esta la mielinizacion preservada por lo que las latencias distales, la velocidad de conduccion no se ven afectadas. Despues cuando el daño axonal es muy importante , se produce una desmielinizacion y se alteran estos parametros. La neuropatia desmielinizante clasicamente cuasa enlentecimiento de la conduccion nerviosa. La amplitud esta preservada. Las neuropatias desmielinizantes hereditarias clasicamente producen anomalias simetricas de la conduccion. Otros procesos donde se produzca desmielinizacion en parche como CIDP pueden generar bloqueos asimetricos en la conduccion. Fig. 4. CMAP. These responses were recorded from the extensor digitorum brevis muscle of the foot following stimulation of the peroneal nerve. The top waveform was recorded after stimulation at the ankle, the middle waveform after stimulation below the knee (inferolateral to the fibular head) and the bottom waveform after stimulation at the knee (in the popliteal fossa). The sweep speed is 5 msec/division and the sensitivity is 5 mV/division. These waveforms have onset latencies of 4.0, 11.1, and 13 msec, respectively (corresponding to the increasing distance between the stimulating and recording electrodes at each of the stimulation sites). Conduction velocities (calculated using latency and inter-electrode distances) are 46 m/sec for the proximal and distal segments of the nerve, whereas the amplitudes are 11.5, 10.4, and 10.2 mV, respectively, all of which are within normal limits. Desmielinizante Axonal The Electrodiagnosis of Neuropathy : Basic Principles and Common Pitfalls. Neurol Clin 25 (2007) 1–28.
 and the bottom waveform after stimulation at the knee (in the popliteal fossa). The sweep speed is 5 msec/division and the sensitivity is 5 mV/division. These waveforms. have onset latencies of 4.0, 11.1, and 13 msec, respectively (corresponding to the increasing distance. between the stimulating and recording electrodes at each of the stimulation sites). Conduction. velocities (calculated using latency and inter-electrode distances) are 46 m/sec for the. proximal and distal segments of the nerve, whereas the amplitudes are 11.5, 10.4, and mV, respectively, all of which are within normal limits. Desmielinizante. Axonal. The Electrodiagnosis of Neuropathy : Basic Principles and Common Pitfalls. Neurol Clin 25 (2007) 1–28.")
6
Electromiografía 1.Actividad de inserción 2.Actividad espontánea
3. PAUM 4. Reclutamiento de UM Durante la EMG se evaluan 5 parametros: Actividad de inserción: es la actvidad que genera la inserción de la aguja en el músculo. Es marcadora de denervacion del musculo, tambien se puede ver en miositis. Actividad espontánea: es la despolarizacion espontánea que tiene el musculo cuando esta en reposo (fibriliaciones y ondas positivas). En individuos sanos no debe existir, ya que esto traduce actividad de denervacion por daño de la neurona motora o su raiz. Tambien se puede ver en polimiosistis. PAUM: activación de la unidad motora. Cuando se produce daño añonal, las fibras que quedan intentan reinervar el musculo, es un proceso que dura meses, y se llama reinervacion colateral. Esto lleva a que un axon inerve varias fibras musculares, eso se traduce en un PAUM mas grande y es lo que se llamara PAUM neurogénico. Esto es marcador de injuria motora cronica. Reclutamiento de unidades motoras En el daño axonal la actividad de insercion y la actividad espontanea aparece 3 semanas despues de la injuria. Los PAUM neurogenicos no apareceran hasta los 2 a 3 meses, que es lo que se requiere para lograr la reinervacion colateral. La desmielinizacion solamente producira cambios en el reclutamiento de unidad motora. Axonal Desmiel The Electrodiagnosis of Neuropathy : Basic Principles and Common Pitfalls. Neurol Clin 25 (2007) 1–28.
. En individuos sanos no debe existir, ya que esto traduce actividad de denervacion por daño de la neurona motora o su raiz. Tambien se puede ver en polimiosistis. PAUM: activación de la unidad motora. Cuando se produce daño añonal, las fibras que quedan intentan reinervar el musculo, es un proceso que dura meses, y se llama reinervacion colateral. Esto lleva a que un axon inerve varias fibras musculares, eso se traduce en un PAUM mas grande y es lo que se llamara PAUM neurogénico. Esto es marcador de injuria motora cronica. Reclutamiento de unidades motoras. En el daño axonal la actividad de insercion y la actividad espontanea aparece 3 semanas despues de la injuria. Los PAUM neurogenicos no apareceran hasta los 2 a 3 meses, que es lo que se requiere para lograr la reinervacion colateral. La desmielinizacion solamente producira cambios en el reclutamiento de unidad motora. Axonal. Desmiel. The Electrodiagnosis of Neuropathy : Basic Principles and Common Pitfalls. Neurol Clin 25 (2007) 1–28.")
7
Neuropatía vs Neuronopatía
La distinción clínica y neurofisiológica no está clara Etiologías se superponen Hipotonía, debilidad, arreflexia, EMG con hallazgos neurogénicos ¿Neuropatía o Neuronopatía?

8
Alteración del transporte axonal
CMT2 dHMN ELA juvenil SPG AME DAÑO AXONAL Alteración del transporte axonal

9
Función axonal Transporte axonal
El transporte intracelular de proteínas y organelos es una función esencial de todas las cl mamíferas, y es especialmente importante en el caso de la neurona. En las neuronas todas las proteínas que se producen en el soma deben ser transportadas por el axón y ubicadas en compartimientos específicos del axón. De esta manera la arquitectura de las neuronas se hace especialmente dependiente del transporte intracelular. Role of Axonal Transport in Neurodegenerative Diseases∗ Kurt J. De Vos,1. Annu. Rev. Neurosci :151–173

10
Transporte axonal La maquinaria para el transporte axonal tiene dos componentes. Motores moleculares Rieles –> los microtúbulos por donde los motores moleculares corren. Role of Axonal Transport in Neurodegenerative Diseases∗ Kurt J. De Vos,1. Annu. Rev. Neurosci :151–173

11
Motores moleculares ATP Transporte anterogrado Transporte retrógrado
ATPasa Transporte anterogrado Transporte retrógrado La velocidad del transporte axonal ira en función del las moléculas que se transportan Clásicamente el transporte axonal se divide según su velocidad: -Transporte rápido: la velocidad es de 1mcm/s , y se transportan mitocondrias y vesículas -Transporte lento: a 1mm/día, que trasporta los componentes del citoesqueleto. Role of Axonal Transport in Neurodegenerative Diseases∗ Kurt J. De Vos,1. Annu. Rev. Neurosci :151–173

12
Transporte axonal y enfermedades neurodegenerativas
La acumulación de organelos o proteínas en el axón o en el soma son el sello patológico de las enfermedades neurodegenerativas. Actualmente se sabe que el daño del transporte axonal es la base fisiopatológica de estas enfermedades. Role of Axonal Transport in Neurodegenerative Diseases∗ Kurt J. De Vos,1. Annu. Rev. Neurosci :151–173

13
Transporte axonal y enfermedades neurodegenerativas
Los mecanismos por lo cuales el transporte axonal se altera son 4: Daño de los “motores” (dineina y kinesina) Daño de los rieles (microtúbulos) Daño de las cargas, que impidan acoplarse a los motores Daño al “combustible de los motores” ATP (mitocondria) Role of Axonal Transport in Neurodegenerative Diseases∗ Kurt J. De Vos,1. Annu. Rev. Neurosci :151–173
 Daño de los rieles (microtúbulos) Daño de las cargas, que impidan acoplarse a los motores. Daño al combustible de los motores ATP (mitocondria) Role of Axonal Transport in Neurodegenerative Diseases∗ Kurt J. De Vos,1. Annu. Rev. Neurosci :151–173.")
14
Transporte axonal y enfermedades neurodegenerativas
Daño en los “motores” Role of Axonal Transport in Neurodegenerative Diseases∗ Kurt J. De Vos,1. Annu. Rev. Neurosci :151–173

15
Transporte axonal y enfermedades neurodegenerativas
Daño en los “rieles” Role of Axonal Transport in Neurodegenerative Diseases∗ Kurt J. De Vos,1. Annu. Rev. Neurosci :151–173

16
Transporte axonal y enfermedades neurodegenerativas
Daño en las “cargas” Role of Axonal Transport in Neurodegenerative Diseases∗ Kurt J. De Vos,1. Annu. Rev. Neurosci :151–173

17
Transporte axonal y enfermedades neurodegenerativas
Daño en el “ proveedor energético” Role of Axonal Transport in Neurodegenerative Diseases∗ Kurt J. De Vos,1. Annu. Rev. Neurosci :151–173

18
Transporte axonal y mielina
Muchos estudios han demostrado relación entre la mielinización y el transporte axonal. Los mecanismos no son bien entendidos pero se cree que la mielina induce señales de fosforilación de los neurofilamentos que están involucrados con el transporte axonal. Many studies have demonstrated a link between myelination and axonal transport (e.g., deWaegh & Brady 1990, deWaegh et al. 1992, Sanchez et al. 2000). Although the mechanisms are not fully understood, myelin probably signals to induce changes to phosphorylation of a number of axonal proteins associated with transport. Neurofilaments are one such protein (deWaegh et al. 1992, Garcia et al. 2003). Defective myelination also causes some forms of Charcot-Marie-Tooth (CMT) disease
. Although the mechanisms. are not fully understood, myelin probably signals. to induce changes to phosphorylation of. a number of axonal proteins associated with. transport. Neurofilaments are one such protein. (deWaegh et al. 1992, Garcia et al. 2003). Defective myelination also causes some forms. of Charcot-Marie-Tooth (CMT) disease.")
19
AME CMT dHMN ELA SPG

20
Charcot-Marie-Tooth Trastorno genéticamente heterogéneo con un fenotipo clínico común Prevalencia 0,08% , 40/ Se afectan nervios motores y sensitivos Los casos típicos se presentan las primeras 2 décadas de la vida y son de curso progresivo La edad de inicio, curso de la enfermedad, velocidad de progresión y la severidad van a depender del tipo de CMT ACTA NEUROLOGICA SCANDINAVICA Volume 126 November Supplement 193 Genetic Epidemiology of Charcot–Marie–Tooth Disease. G. J. Braathen

21
Cuadro clínico Tienen un sello diagnóstico que permite diferenciarlas
Varían según edad de inicio Todas presentan debilidad y atrofia distal progresiva (variable), simétrica, arreflexia Inicio en EEII después progresa a EESS Dificultad en la marcha marcha en steppage Dificultad para manipular objetos Pies fries, acrocianosis. Temblor de manos Compromiso sensitiva variable en magnitud y modalidad parestesias Deformidad en los pies (pie cavo, dedos en martillo Tienen un sello diagnóstico que permite diferenciarlas Diagnosis, natural history, and management of Charcot–Marie–Tooth disease. Lancet Neurol 2009; 8: 654–67
, simétrica, arreflexia. Inicio en EEII después progresa a EESS. Dificultad en la marcha marcha en steppage. Dificultad para manipular objetos. Pies fries, acrocianosis. Temblor de manos. Compromiso sensitiva variable en magnitud y modalidad parestesias. Deformidad en los pies (pie cavo, dedos en martillo. Tienen un sello diagnóstico que permite diferenciarlas. Diagnosis, natural history, and management of Charcot–Marie–Tooth disease. Lancet Neurol 2009; 8: 654–67.")
22
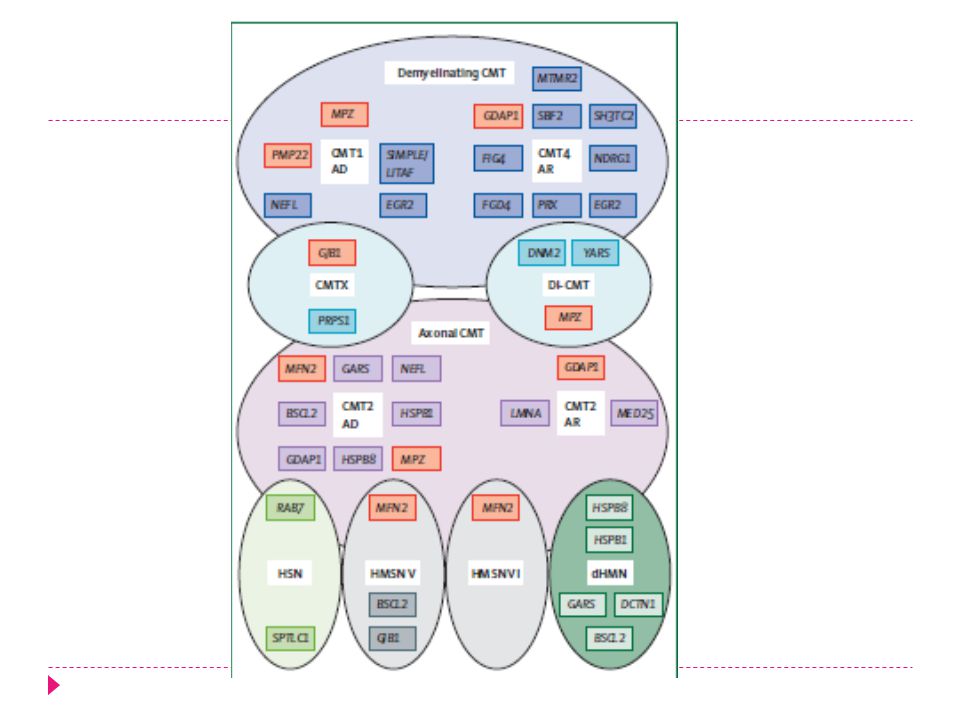
Clasificación CMT Cuadro clinico Electrofisiologia CMT 2 Axonal
Desmielinizante CMT 1 Historia familiar Dominante Recesivo Ligado a X Esporadico

23
Clasificación Desmielinizante Dominante CMT1 Axonal CMT2
Dominante ligX CMT3 Recesiva CMT4 CMT4C

25
CMT 1 CMT1A es el mas frecuente Duplicación PMP 22.
Usualmente se presenta con el fenotipo clásico Velocidad de conducción alterada Son de curso “benigno”

26
CMT 2 CMT2 menos frecuente que CMT1 Prevalencia 1-40/100.000
20% de todos los pacientes con CMT.

28
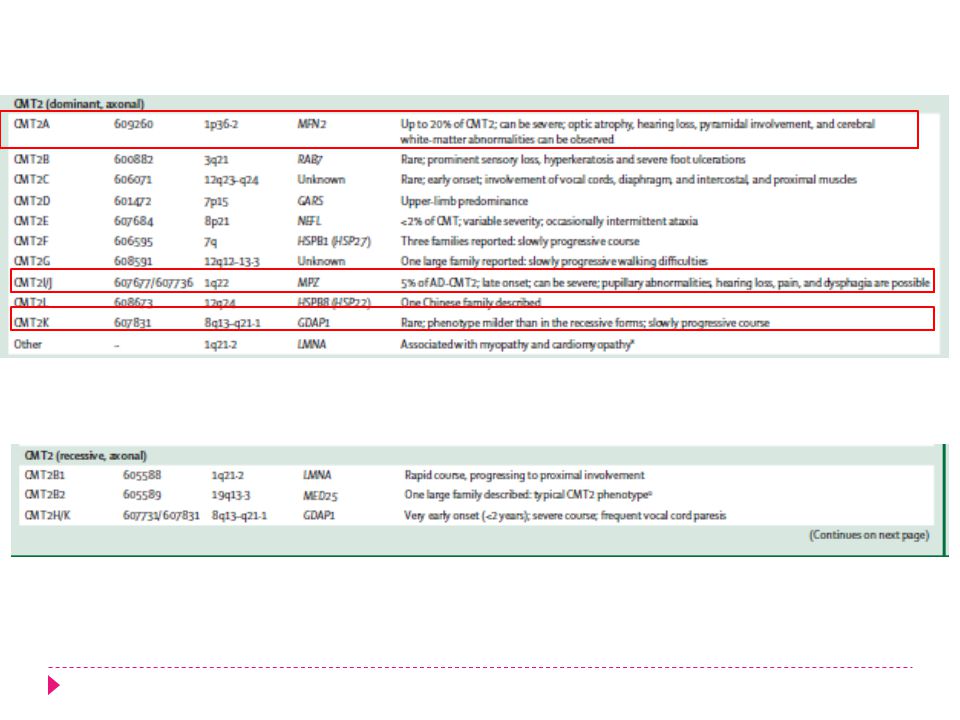
CMT 2 GEN PROTEINA SELLO DG CMT2A Mitofusina KIF1B Kinesina 1
Compromiso ES. Piramidalismo. Atrofia óptica. CMT2B Rab7 GTPasa Rab7 Edad años Severo compromiso sensitivo CMT2C TRPV 4 Canal de calcio Compromiso proximal, diafragma, intercostal y cuerdas vocales CMT2D GARS Glycyl-tRNA sintetasa Compromiso de músculos de la mano. CMT2E NEFL Cadena liviana de Neurofilamentos Inicio < 13 años. Compromiso sensitivo en todas modalidades.

29
CMT 2 GEN PROTEINA SELLO DG CMT2F HSPB1 Heat Shock protein 1
Inicio > 15 años. Lentamente progresiva CMT2G ? Lenta progresion. Preserva ROT rotuliano. *CMT2H GDAP1 Lentamente progresiva. Parálisis de cuerda vocal. Atrofia óptica. CMT2I/J MPZ Inicio tardío años. Pupila de Adie. Sordera. CMT2L HSPB8/22 Minimo compromiso sensitivo. Esocliosis. Charcot-Marie-Tooth Disease: A Clinico-genetic confrontation. Annals of Human Genetics (2008) 72,416–441
 72,416–441.")
30
CMT2A: Mitofusina 2 La mitofusina es una GTPasa de transmembrana.
Presente en la membrana mitocondrial externa Regula la arquitectura mitoncodrial. Indispensable para mantener la integridad estructural de la mitocondria y la movilidad mitocondrial dentro de la neurona Role of mitofusin 2 mutations in the physiopathology of Charcot–Marie–Tooth disease type 2A Romain Cartoni, Jean-Claude Martinou ⁎. Experimental Neurology 218 (2009) 268–273
 268–273.")
31
Rol de la Mitofusina 2

32
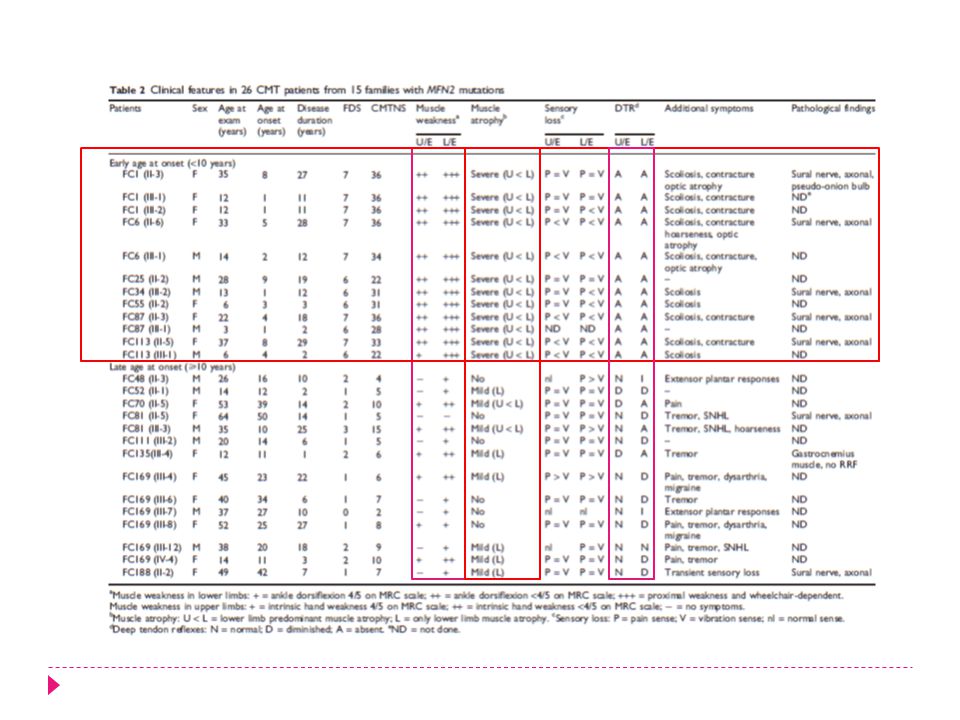
N:218 62 familias con CMT2 15 familias CMT2A (24%) N: H y 15 M Mutación de novo en 5 familias (33.3%)
 .")
33
N:218 62 familias con CMT2 15 familias CMT2A (24%) N: H y 15 M Mutación de novo en 5 familias (33.3%) Existe una relación genotipo - fenotipo

34
Presentación clínica Debilidad y atrofia de inicio distal
Inicio Precoz < 10 años 3,9 años (1-9 años) Debilidad y atrofia de inicio distal Inicial en EEII después EESS Paresia distal desde leve hasta plejia Compromiso sensitivo 92,3% Debilidad severa + arreflexia Mayor compromiso de EESS Deformidades en los pies Abatiestesia 66% Escoliosis y contracturas 38,5% Atrofia óptica 10-20% Inicio Tardío > 10 años años (10-50 años) Debilidad moderada Deformidades de pies leves Abatiestesia 21% Temblor postural y dolor 30% Sordera SN 11%
 Debilidad y atrofia de inicio distal. Inicial en EEII después EESS. Paresia distal desde leve hasta plejia. Compromiso sensitivo 92,3% Debilidad severa + arreflexia. Mayor compromiso de EESS. Deformidades en los pies. Abatiestesia 66% Escoliosis y contracturas 38,5% Atrofia óptica 10-20% Inicio Tardío. > 10 años 23.9 años (10-50 años) Debilidad moderada. Deformidades de pies leves. Abatiestesia 21% Temblor postural y dolor 30% Sordera SN 11%")
36
Electrofisiología Todos presentan compromiso axonal, predominio motor, distal y de EEII Existe una correlación entre la debilidad y la amplitud del (CMAP) No existe correlación entre la VC y la debilidad distal Inicio Precoz < 10 años 3,9 años (1-9 años) Debilidad severa + arreflexia Compromiso mas severo, menores amplitudes Inicio Tardío > 10 años años (10-50 años) En el grupo precoz las amplitudes del CMAP eran menores que en el grupo de inicio mas tardio. En el grupo de inicio tardio las velocidades de conduccion fueros normales. Esto demuesttra que la degeneracion axonal progresa mas rapido en pacientes con inicio tardio. Debilidad moderada > Compromiso sensitivo
 No existe correlación entre la VC y la debilidad distal. Inicio Precoz. < 10 años 3,9 años (1-9 años) Debilidad severa + arreflexia. Compromiso mas severo, menores amplitudes. Inicio Tardío. > 10 años 23.9 años (10-50 años) En el grupo precoz las amplitudes del CMAP eran menores que en el grupo de inicio mas tardio. En el grupo de inicio tardio las velocidades de conduccion fueros normales. Esto demuesttra que la degeneracion axonal progresa mas rapido en pacientes con inicio tardio. Debilidad moderada. > Compromiso sensitivo.")
37
RNM 8/21 presentaron alteraciones en RNM 7 de inicio tardío
Presencia de síntomas asociados, sordera, disartria, piramidal

38
Histopatología Se realizo biopsia en 7 pacientes.
Perdida de fibras largas mielinizadas en pacientes de inicio precoz. Fibras atroficas y agrupadas Formaciones bulbares “pseudo-onion”

39
AME “clásico” Debilidad de tronco y proximal > distal, inferior>superior Simétrica Fasciculaciones en la lengua Ausencia del gen SMN 1 Molecular Mechanisms of Spinal Muscular Atrophy. J Child Neurol 2007; 22; 979

40
AME AME es la consecuencia de la mutación del gen SMN 1.
Este gen tiene 2 funciones: Ensamblaje y regeneración de las ribonucleinas encargadas del splicing Transporte del RNA mensajero a través del axón. La mayoría de las proteínas son sintentizadas en el soma de la neurona, pero hay un grupo que son sintetizadas en el axón gracias al transporte del RNAm. Una de las proteínas sintetizadas es la B-actina que es responsable del crecimiento del cono axonal.

41
Variantes AME 63 pacientes 48 AME “clásico” 15 AME “atípico”
Inclusión: Debilidad muscular progresiva y atrofia Inicio en la infancia EMG con patrón neurogenico Estudio genetico SMN 1+

42
Hand tremor was reported in 46% of typical SMA
cases (11 types II, 6 types III, and 5 types I) while it was observed in only 13% of atypical SMA cases. Tongue fasciculations were found in 64% of SMA type I and only in 15% of SMA type II, 33% of SMA type III, and 6.5% of atypical SMA cases (patient 5). We did not find any case of myocardic dysfunction or extra-ocular muscle involvement nor sensory disturbance in the 2 groups. All cases of arthrogryposis were seen in the atypical SMA group (with one case of congenital fracture: patient 14). One case of SMA type I had a right phrenic palsy. Four typical SMA presented marked facial weakness (2 upper facial palsies, one palpebrae orbicular paresis and one asymmetrical facial palsy
 while it. was observed in only 13% of atypical SMA cases. Tongue fasciculations were found in 64% of SMA. type I and only in 15% of SMA type II, 33% of SMA. type III, and 6.5% of atypical SMA cases (patient 5). We did not find any case of myocardic dysfunction or. extra-ocular muscle involvement nor sensory disturbance. in the 2 groups. All cases of arthrogryposis were seen in the atypical. SMA group (with one case of congenital fracture: patient 14). One case of SMA type I had a right phrenic palsy. Four typical SMA presented marked facial weakness. (2 upper facial palsies, one palpebrae orbicular paresis. and one asymmetrical facial palsy.")
44
AME con distress respiratorio con la mutación en el gen IGHMBP2 (11q).
AME con hipoplasia pontocerebelosa AME con epilepsia mioclónica progresiva AME con artrogriposis neurogénica y fracturas congénitas AME distal AME escapuloperoneal (chromosome 12q24) AME con DR, tienen paralisis diafragmatica, con insuficiencia respiratoria temprana y muerte precoz
 AME con DR, tienen paralisis diafragmatica, con insuficiencia respiratoria temprana y muerte precoz.")
45
Non-5q spinal muscular atrophies. Neurology 2011;77:312–314

46
Neuropatías motoras distal hereditarias
Grupo heterogéneo de causa genética. 80% aún sin genes identificados Compromiso exclusivamente motor, aunque hay algunas descritas con compromiso sensitivo. También conocidas como AME distal Clínica: Inicio antes de los 20 años Curso progresivo lento Atrofia y debilidad distal, arreflexia Inicio en EEII después en EESS afectando musculatura interósea Compromiso bulbar es raro Solo compromiso motor EMG denervación distal crónica The distal hereditary motor neuropathies .Alexander M Rossor,. J Neurol Neurosurg Psychiatry 2012;83:6e14

47
Clasificación The distal hereditary motor neuropathies .Alexander M Rossor,. J Neurol Neurosurg Psychiatry 2012;83:6e14

48
HSPB1 y HSPB8, que son proteínas chaperonas.
Importantes en la respuesta celular al stress. Ambas interactúan con varios filamentos para la formación de nuevos neurofilamentos, La mutación produce alteración en el ensamblaje de los neurofilamentos, y por lo tanto no podrán transportarse por el axón.

49
CMT 2F CMT 2L CMT 2D CMT 2C ELA J

50
ELA juvenil ELA “clásico” es la enfermedad de motoneurona mas frecuente. 90% esporádicas multifactorial 10% familiar genética ELA juvenil inicio antes de los 25 años Signos de 1MN: Babinski, clonus, espasticidad Signos de 2°MN: Atrofia, debilidad, fasciculaciones Sin compromiso sensitivo Pocas familias reportadas. Curso lentamente progresivo Compromiso facial y bulbar Inicio < 6 años.sin comp bulbar Atrofia de manos, pies, lengua y faringe Juvenile amyotrophic lateral sclerosis. Handbook of Clinical Neurology, Vol. 82 (3rd series).2007
")
51
Paraparesia espástica hereditaria
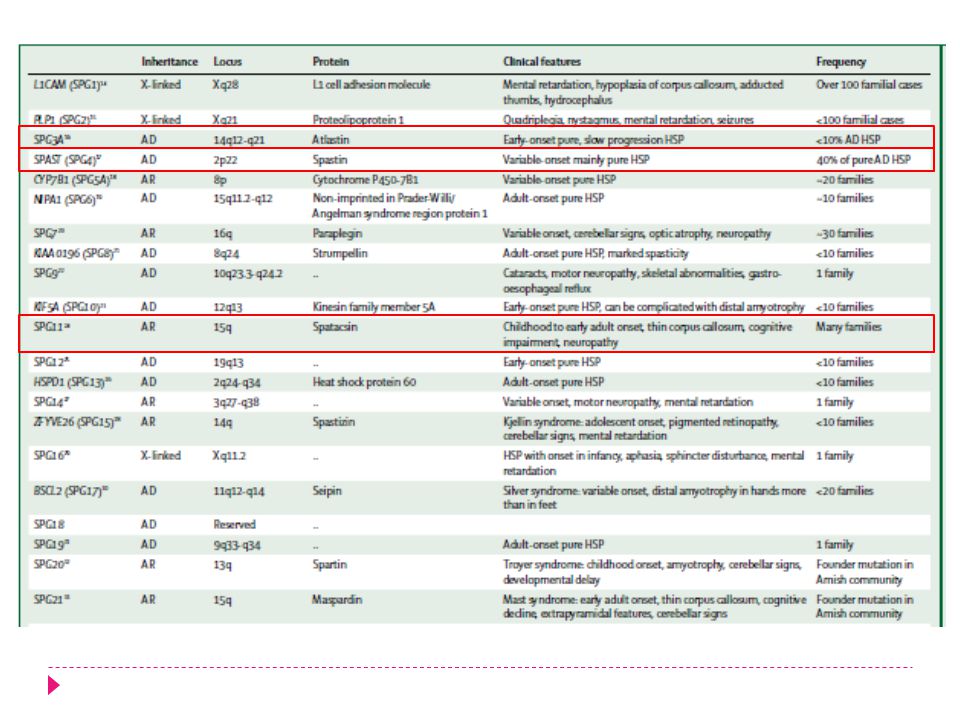
Grupo heterogéneo de enfermedades que afectan la primera motoneurona. Se han identificado 15 mutaciones responsables, cuyo mecanismo patogénico final es la alteración en el transporte axonal. 4 mecanismos fisiopatológicos: Alteración de la kinesina Alteración de los microtubulos Daño mitocondrial Daño de mielinización

52
Alteración de la kinesina
Daño Axonal en SPG Alteración de la kinesina Alteración de los microicrotubulos SPG 4 SPG 3A 40% SPG 10 SPG 1 SPG 2 Daño mitocondrial Daño mielinización SPG 7

53
Paraparesia Espástica Hereditaria
SPG Dominante Recesiva Ligada X Clínica: Edad de inicio variable Historia familiar Inicio en EEII, espasticidad sin o mínima paresiadificultad para caminar Urgencia urinaria Compromiso sensitivo variable “plus” o compleja si se asocian otras manifestaciones neurológicas

56
Neuropatía axonal por Enfermedad celíaca

57
Resumiendo… CMT - / + -/+ ++ - ELA + +/- AME AME d SPG +++
Debilidad distal Debilidad proximal Reflejos Fasciculasciones Alteración Sensibilidad Compromiso esfinter Compromiso bulbar Compromiso SNC CMT - / + -/+ ++ - ELA + +/- AME AME d SPG +++

58
En suma…. El daño del transporte axonal es producido por mecanismos complejos y es la base fisiopatológica de las enfermedades neurodegenerativas. Los genes implicados en estas patologías se comparten haciendo cada vez más difícil la diferenciación clínica de estas entidades.

59
Caso clínico Cuadro clínico de inicio en la infancia, PROGRESIVO.
Compromete tronco y 4 ext Debilidad distal > proximal Deformidad en los pies Compromiso sensitivo Fasiculaciones linguales? Signos piramidales Deterioro cognitivo? Compromiso motor y sensitivo Daño axonal ¿Compromiso SNC? No hay antecedentes familiares

60
CMT - / + -/+ ++ - ELA + +/- AME AME d SPG +++ CMT 2A SPG
Debilidad distal Debilidad proximal Reflejos Fasciculasciones Alteración Sensibilidad Compromiso esfinter Compromiso bulbar Compromiso SNC CMT - / + -/+ ++ - ELA + +/- AME AME d SPG +++ CMT 2A SPG

61
CMT 2A Porqué si Porqué no Porque sí Porque no SPG “PLUS” AR SPG 11
Frecuencia 1/3 son esporádicos Compromiso sensitivo y motor Deterioro cognitivo leve Pueden tener presencia de reflejos Porqué no Marcha previa “paraparética” Porque sí Presencia de reflejos Marcha previa “paraparética” Deterioro cognitivo leve Algunas tienen neuropatía (SPG 11) Porque no Mucho compromiso sensitivo Fasciculaciones linguales
 Porque no. Mucho compromiso sensitivo. Fasciculaciones linguales.")
62
CAUSAS GENÉTICAS DEL COMPROMISO AXONAL
Dra. Susana Lara Dra. Alejandra Méndez Neuróloga Infantil Residente de Neurología Infantil ENERO 2014

Presentaciones similares