Descargar la presentación
La descarga está en progreso. Por favor, espere

1
USO SEGURO DE MEDICAMENTOS FARMACOCINETICA CLINICA
JULIO CESAR GARCIA CASALLAS QF MD Msc DEPARTAMENTO DE FARMACOLOGIA CLINICA Y TERAPEUTICA

4
FARMACOCINÉTICA FARMACODINÁMIA TEJIDO PLASMA BIOTRANSFORMACIÓN
ELIMINACIÓN ORINA HECES/BILIS ADMINISTRACIÓN L A D M E FARMACODINÁMIA UNIÓN RECEPTOR EFECTO TOXICO EFECTO TERAPÉUTICO DEGRADACIÓN INTRACELULAR COMPETITIVA [RECEPTOR]

5
FARMACOCINÉTICA Es la rama de la Farmacología que estudia el paso del Fármaco a través del organismo en función del tiempo y de la dosis. Comprende los procesos de liberación, absorción, distribución, metabolismo o biotransformación y excreción de los Fármacos.

6
LADME FARMACOCINETICA
Es el area de la Farmacología que estudia el recorrido y las modificaciones que experimentan los fármacos y su metabolitos en el interior del organismo. IBERACION BSORCION PROCESO FARMACOLOGICO LADME ISTRIBUCION ETABOLISMO XCRECION

7
ABSORCION EXCRECION LIBERACION SITIO DE ACCION TEJIDO RECEPTOR
METABOLISMO SITIO DE ACCION RECEPTOR FARMACO LIBRE FARMACO LIGADO TEJIDO FARMACO LIBRE FARMACO LIGADO EXCRECION

8
Farmacocinética

9
CICLO FARMACOCINÉTICO
Administración Liberación Absorción (membrana(s)) Excreción/ Metabolismo Fármaco en sangre Distribución Distribución Intensidad y duración de la respuesta [Fármaco en el sitio de acción]
) Excreción/ Metabolismo. Fármaco en sangre. Distribución. Distribución. Intensidad y duración de la respuesta. [Fármaco en el sitio de acción]")
10
LIBERACION Los medicamentos se expenden bajo una determinada forma de presentación :

11
LIBERACION DESINTEGRACION DESAGREGACION DISOLUCION GRANULOS CAPSULAS
TABLETAS DESAGREGACION PARTICULAS SOLUCIONES DISOLUCION

12
Ciclo farmacocinético
GI Otras vías SANGRE Sitios de acción Depósi-tos Absorción Distribución Eliminación

13
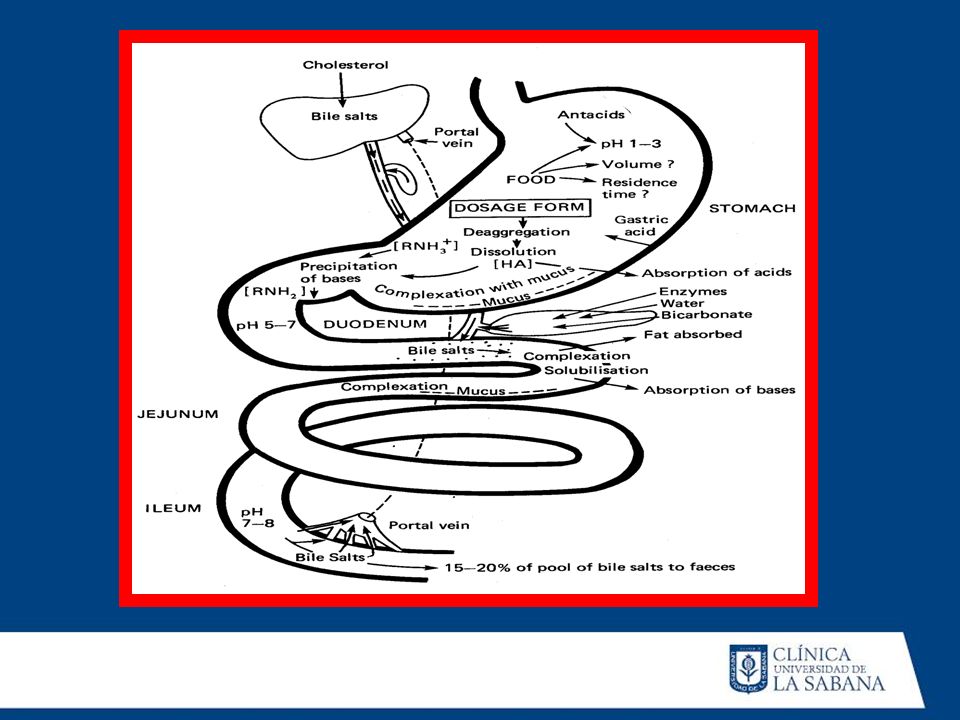
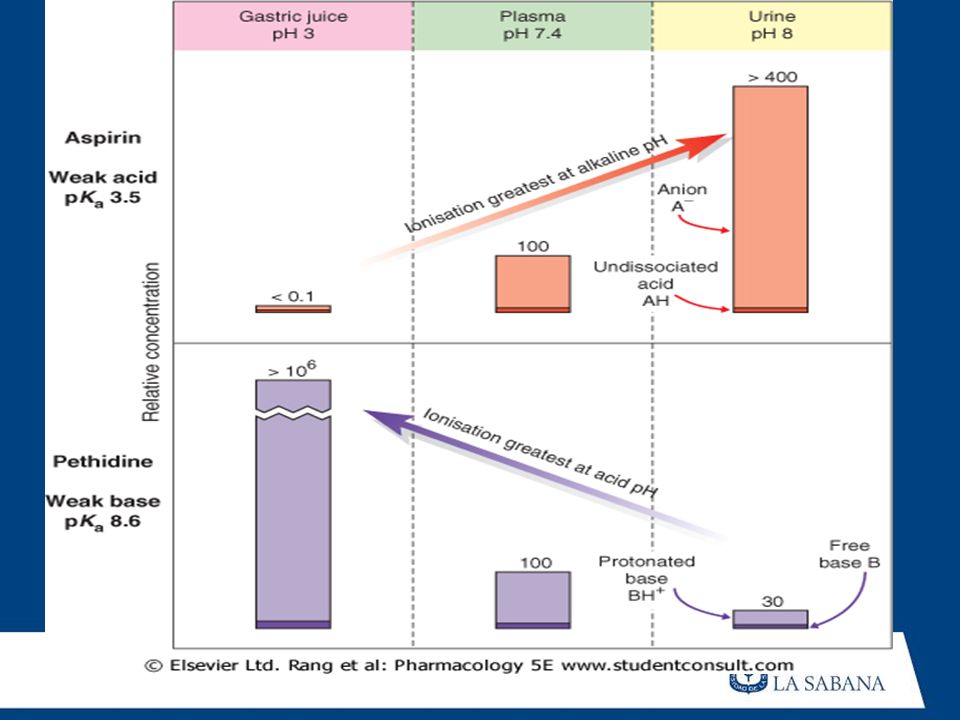
ABSORCION Es el pasaje del Fármaco desde el sitio de aplicación hacia el interior del organismo (CIRCULACION SANGUINEA). ZONA DE ABSORCION : lugar donde el fármaco ingresa a la circulación VIA DE ADMINISTRACION : indica el lugar por donde se suminitra el fármaco. No siempre coinciden con la zona de absorción IMPORTANCIA DE LA VELOCIDAD DE ABSORCION : La velocidad de la absorción permite determinar : La Vía de administración: más rápida via EV, más lenta VO La Dosis : menor dosis menor absorción La rapidez del inicio de acción: vía EV más rápida acción

14
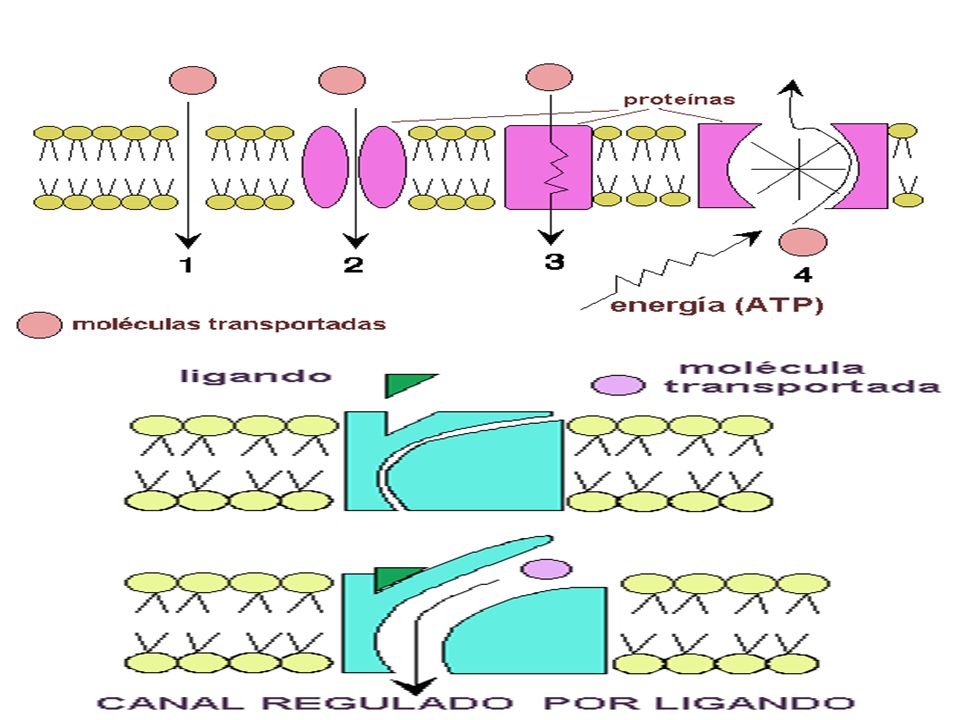
MECANISMOS DE ABSORCION
PROCESOS PASIVOS: siguiendo las Leyes físicas de gradientes de concentración, de potencial o de Presión Hidrostática. No hay consumo de Energia *DIFUSION SIMPLE: Na y K *DIFUSION FACILITADA: Insulina-Glucosa *FILTRACION : en riñón. Paso de solutos a través de membranas siguiendo leyes de FICK Y HENDERSON HASSELBACH, por diferencia de presión Hisrostática. * OSMOSIS : es el pasaje de solventes a través de una membrana por diferencia de presión Osmótica.

15
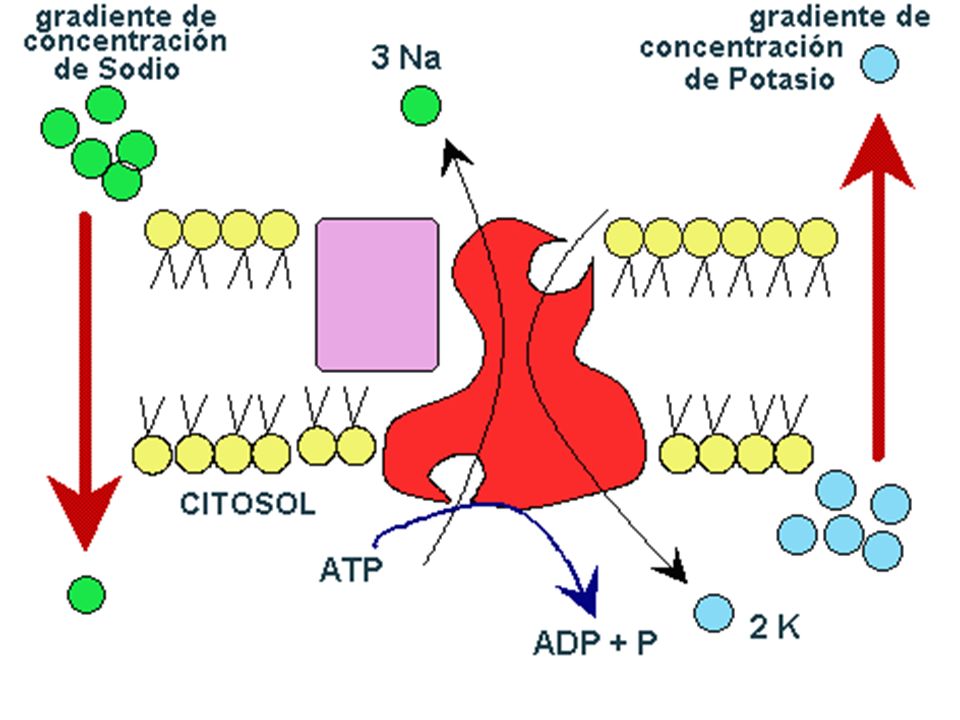
MECANISMOS DE ABSORCION
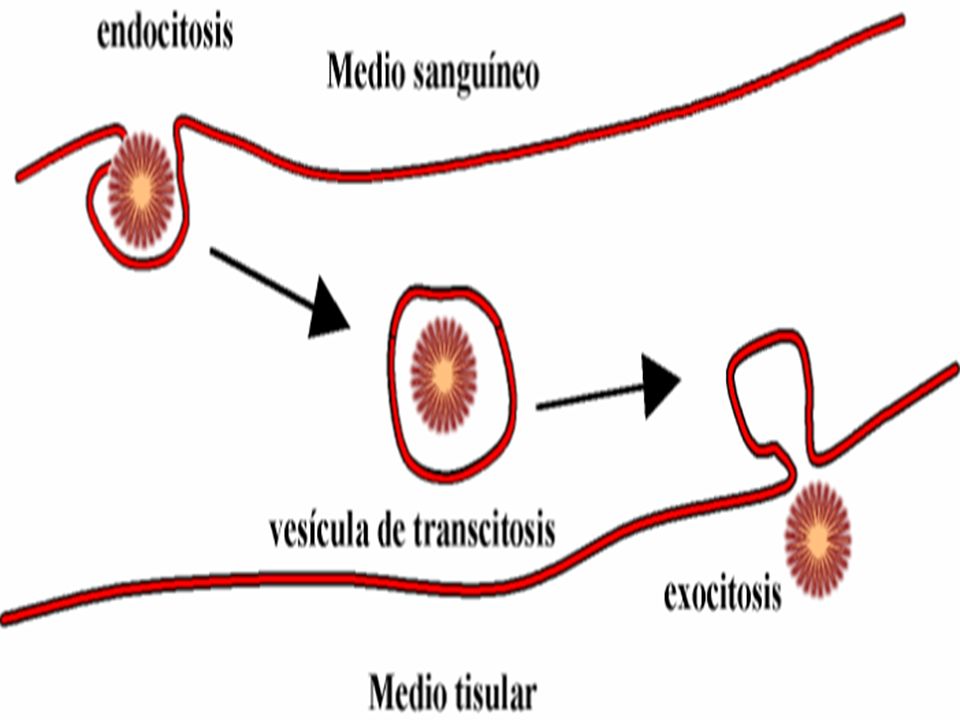
2.- TRANSPORTE ESPECIALIZADO : existe gasto de energía y puede ser de 2 clases : TRANSPORTE ACTIVO : se realiza contra las gradientes de concentración : Bombas de Na+ y K+, Ca++ y Mg++. Se caracteriza por ser saturable, es decir a partir de una determinada concentración no aumenta la velocidad de transporte. PINOCITOSIS o VESICULACION : Endocitosis o Exocitosis . Se emplea para las proteínas o macromoléculas con PM mayor a 1,000 mmol. Ejm : Pasaje de Fierro y Vitamina B12 del Intestino a la sangre

16
MECANISMOS DE ABSORCION

17
DIFUSION

21
Pinocitosis Fagocitosis

23
FACTORES QUE MODIFICAN LA ABSORCION
A) CARATERISTICAS DE FARMACO: Forma de presentación Tamaño molecular Gradiente de concentración Coeficiente de participación liquido/ agua (Liposolubillidad) Grado de ionización
 CARATERISTICAS DE FARMACO: Forma de presentación. Tamaño molecular. Gradiente de concentración. Coeficiente de participación liquido/ agua (Liposolubillidad) Grado de ionización.")
24
b) Características de la superficie absorbente
Area de absorción : TGI Irrigación TGI : por presencia de alimentos. Factores fisiológicos o patológico : velocidad de vaciamiento gástrico, motilidad intestinal etc. BIODISPONIBILIDAD : Representa la fracción de dosis administrada (por cualquier vía) que alcanza la circulación general en forma inalterada (activa) Via EV : la disponibilidad es de 100% Vía oral : la biodisponivilidad no alcanza el 100% : Efecto de primer paso o metabolismo pre sistémico
 que alcanza la circulación general en forma inalterada (activa) Via EV : la disponibilidad es de 100% Vía oral : la biodisponivilidad no alcanza el 100% : Efecto de primer paso o metabolismo pre sistémico.")
29
DISTRIBUCION : COMPARTIMENTOS IT IC 15% 40% IV 5% FASES :
FASE INICIAL : a los órganos más irrigados SEGUNDA FASE : va zonas regionales COMPARTIMENTOS IT 15% IC 40% IV 5%

30
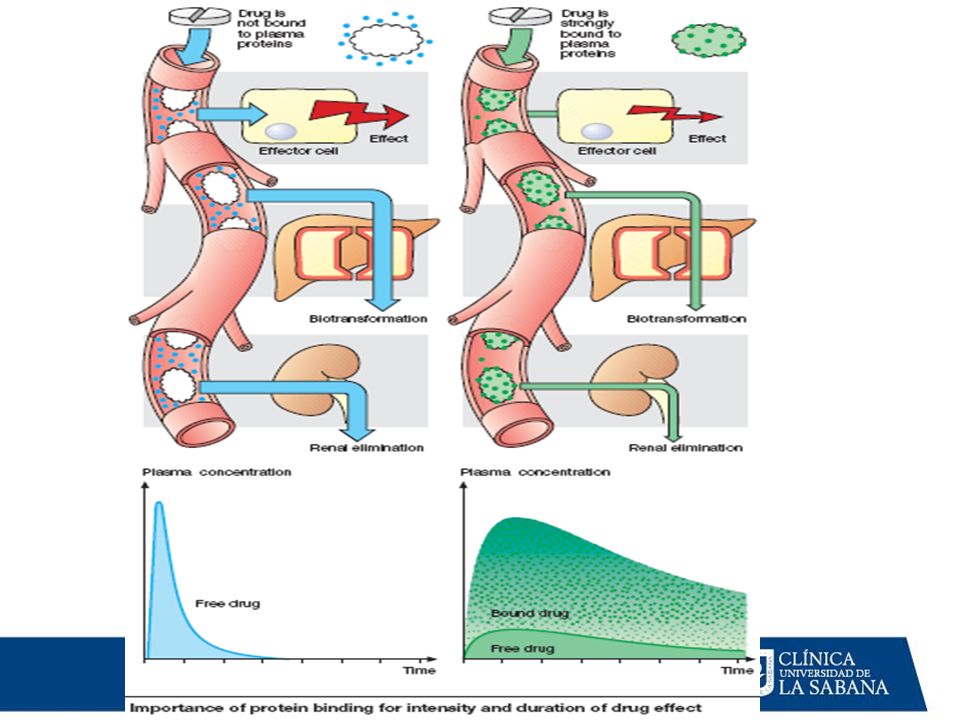
MECANISMOS DE DISTRIBUCIÓN
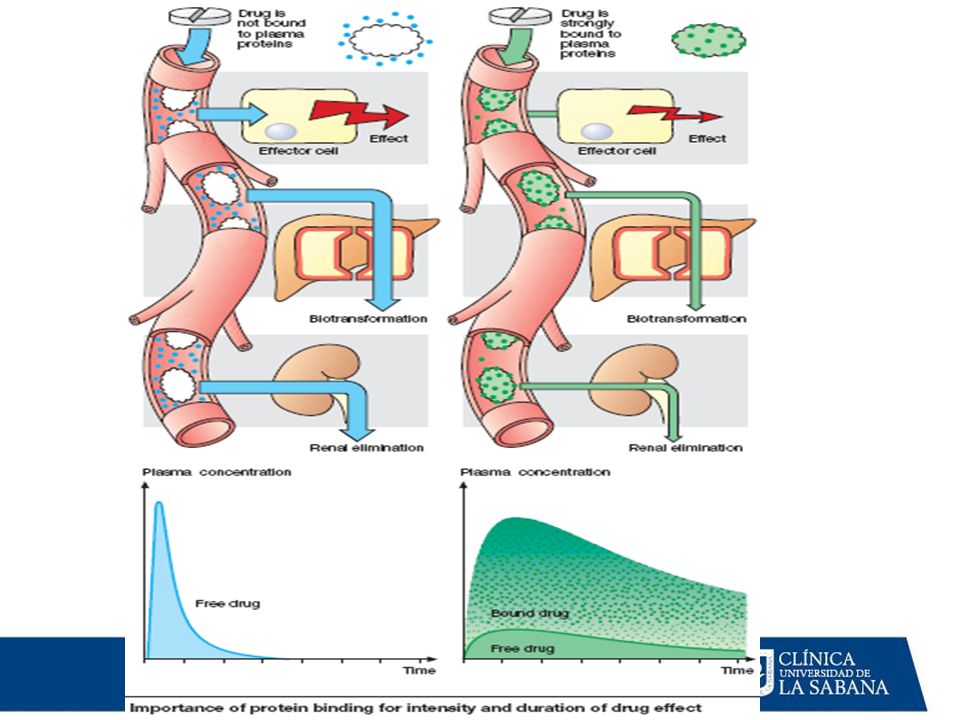
Los fármacos circulan en la plasma en dos formas: Fracción libre: Constituye la fracción farmacológicamente activa, pues es la que puede difundir hasta los tejidos donde ejerce su acción (fracción difusible); sin embargo, por esta misma razón, también está disponible para el metabolismo y excreción pues puede alcanzar los órganos donde se metaboliza y excreta. Fracción ligada a las proteínas plasmáticas: Se halla unida principalmente a las albúminas, por lo cual no puede difundir hacia los tejidos (fracción no difusible): Es la fracción farmacológicmanete inactiva (carece de acción), pues es incapaz de alcanzar los tejidos en donde ejerce sus efectos. Atraviesa poco las membranas, por lo cual: No llega a los tejidos. No cruza la BHE (no alcanza concentraciones adecuadas en el LCR). Se metaboliza con dificultad. Sirve como reservorio del fármaco en la sangre que se libera con lentitud.
; sin embargo, por esta misma razón, también está disponible para el metabolismo y excreción pues puede alcanzar los órganos donde se metaboliza y excreta. Fracción ligada a las proteínas plasmáticas: Se halla unida principalmente a las albúminas, por lo cual no puede difundir hacia los tejidos (fracción no difusible): Es la fracción farmacológicmanete inactiva (carece de acción), pues es incapaz de alcanzar los tejidos en donde ejerce sus efectos. Atraviesa poco las membranas, por lo cual: No llega a los tejidos. No cruza la BHE (no alcanza concentraciones adecuadas en el LCR). Se metaboliza con dificultad. Sirve como reservorio del fármaco en la sangre que se libera con lentitud.")
31
MECANISMOS DE DISTRIBUCION:
FARMACO ALBUMIMNA ALBUMINA

32
TIPOS DE UNION: REVERSIBLE: * ENLACE IONICO O ELECTROVALENT
* ENLACE DE HIDROGENO o VAN DER WAALS o DIPOLAR B) IRREVERSIBLE : ENLACE COVALENTE
 IRREVERSIBLE : ENLACE COVALENTE.")
33
FACTORES QUE MODIFICAN LA DISTRIBUCION
PROPIEDADES FISICO QUIMICAS DEL FARMACO : * Se distribuyen mejor los fármacos Liposolubles, No ionizado y los de Bajo peso molecular GASTO CARDIACO : alcanzan concentraciones elevadas en los órganos más irrigados. PERMEABILIDAD CAPILAR : a mayor permeabilidad mejor distribución de los fármacos. CONTENIDO LIPIDICO DEL TEJIDO : existen fármacos que son muy Liposolubles quedando atrapados en los Tejidos grasos y no aptos para distribución GRADO DE UNION A LAS PROTEINAS PLASMATICAS : Sólo la fracción libre (la no unida a la proteinas) pueden difundir libremente. BARRERAS CORPORALES : *Barrera Hematoencefálica *Barrera Placentaria
 pueden difundir libremente. BARRERAS CORPORALES : *Barrera Hematoencefálica. *Barrera Placentaria.")
34
BARRERA HEMATOENCEFALICA:
BARRERAS CORPORALES BARRERA HEMATOENCEFALICA: * Es permeable a Sustancias Liposolubles e Hidrosolubles muy pequeñas (Urea, Alcohol) * Las sustancias muy Ionizadas son incapaces de pasarlos (Penicilina y Aminas cuaternarias), pero si hay inflamación de las Meninges (MENINGITIS) aumenta la vasodilatación y permite el pasaje de los antibióticos. BARRERA PLACENTARIA *Los fármacos la atraviesan por Difusión pasiva, facilitada y Pinocitosis (Inmunoglobulinas tipo gammaglobulinas) * Es permeable a fracciones no ionizadas, a los no electrolitos liposolubles (cloroformo, Eter y sustancias volátiles) * Es permeable a Hormonas esteroideas, alcohol, salicilatos, atropina, Barbitúricos, antibióticos , alcaloides y antihistaminicos.
 * Las sustancias muy Ionizadas son incapaces de pasarlos (Penicilina y Aminas cuaternarias), pero si hay inflamación de las Meninges (MENINGITIS) aumenta la vasodilatación y permite el pasaje de los antibióticos. BARRERA PLACENTARIA. *Los fármacos la atraviesan por Difusión pasiva, facilitada y Pinocitosis (Inmunoglobulinas tipo gammaglobulinas) * Es permeable a fracciones no ionizadas, a los no electrolitos liposolubles (cloroformo, Eter y sustancias volátiles) * Es permeable a Hormonas esteroideas, alcohol, salicilatos, atropina, Barbitúricos, antibióticos , alcaloides y antihistaminicos.")
35
MODELOS DE DISTRIBUCIÓN
El agua corporal total, que en el adulto representa un 60% del peso corporal total, está distribuido de la siguiente manera: En el espacio intracelular: 40% del peso corporal. En el espacio extracelular: Intravascular (plasma): 5% Intersticial: 15% Por ej, en un sujeto de 70 kg., el agua corporal total será de 42 litros, el agua intravascular 3,5 litros y el agua intersticial alrededor de 10,5 litros. La farmacocinética considera al organismo dividido en compartimiento, acuosos o no, pero siempre, “sectores virtuales del organismo en los cuales el medicamento se considera distribuido uniformemente”.
: 5% Intersticial: 15% Por ej, en un sujeto de 70 kg., el agua corporal total será de 42 litros, el agua intravascular 3,5 litros y el agua intersticial alrededor de 10,5 litros. La farmacocinética considera al organismo dividido en compartimiento, acuosos o no, pero siempre, sectores virtuales del organismo en los cuales el medicamento se considera distribuido uniformemente .")
36
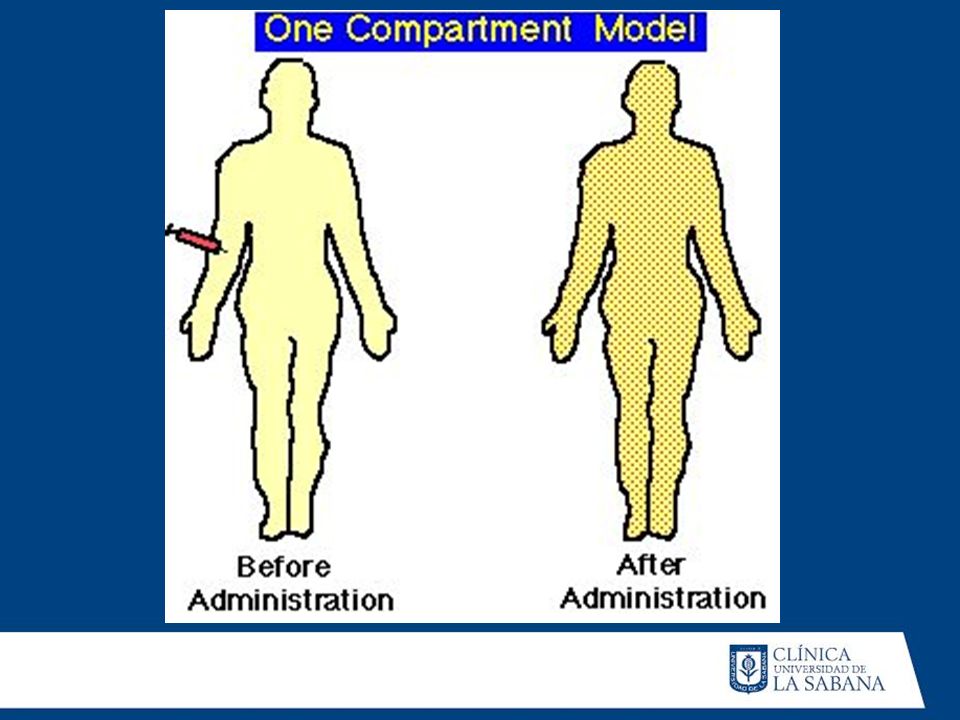
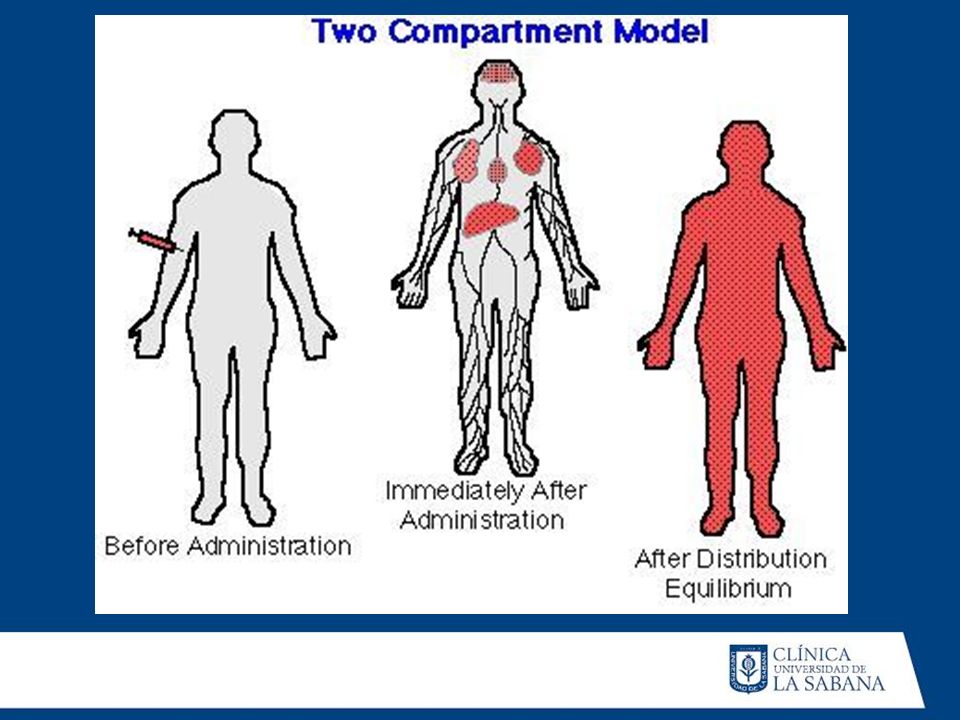
A) MODELO UNICOMPARTAMENTAL
Compartimento Central EXCRECION ABSORCION B) MODELO BICOMPARTAMENTAL ABSORCION Compartimento central Compartimento de 2do orden EXCRECION
 MODELO BICOMPARTAMENTAL. ABSORCION. Compartimento central. Compartimento de 2do orden. EXCRECION.")
37
VOLUMEN DE DISTIBUCION (Vd)
El Vd relaciona la cantidad de fármaco en el organismo con su concentración plasmática: Vd = Cantidad de fármaco en el organizmo (en mg) Concentración plasmática del fármaco (en mg/L)
 Concentración plasmática del fármaco (en mg/L)")
38
Parámetros farmacocinéticos después de una administración VO.
Concentración al pico (Cmax) 4,5 4 Tiempo al pico de la Concentración (tmax) 3,5 3 2,5 Área bajo la curva De la Conc. Vs Tempo (AUC) Concentración plasmática (µg/ml) 2 1,5 Vida media de eliminación (t1/2) 1 0,5 2 4 6 8 10 12 Tiempo después de la administración (h)
 4,5. 4. Tiempo al pico de la. Concentración (tmax) 3, ,5. Área bajo la curva. De la Conc. Vs Tempo. (AUC) Concentración plasmática (µg/ml) 2. 1,5. Vida media de eliminación. (t1/2) 1. 0, Tiempo después de la administración (h)")
39
Parámetros farmacocinéticos después de una administración i.v.
9 8 7 6 Volumen de distribución (Vd) Concentración plasmática (µg/ml) 5 4 3 Depuración Cl 2 t1/2 AUC 1 2 4 6 8 10 12 Tiempo después de la administración (h)
 Concentración plasmática (µg/ml) Depuración. Cl. 2. t1/2. AUC Tiempo después de la administración (h)")
40
BIODISPONIBILIDAD ABSOLUTA
Biodisponibilidad = {AUC oral / AUC i.v. } x 100 Fármaco i.v. AUC i.v. Concentración Plasmática Fármaco p.o. AUC oral Tiempo

41
Biodisponibilidad oral
Fármaco % f Ácido Valproico 100 1 Acetaminofén 88 0.88 Ampicilina 35 0.35 Gentamicina Fenobarbital Ciprofloxaciona 95 0.95

42
Tiempo medio de eliminación, T½ o vida media:
Tiempo necesaria para que la mitad del farmaco sea eliminada del torrente sanguíneo., Tiempo en el cual se elimina el 50 % del farmaco presente en el cuerpo en un momento dado.

44
Importancia clínica de la unión F-P
1.- [ F ] libre actividad farmacológica y clearance renal 2.- Desplazamiento competitivo a.- entre Fármacos b.- por substancias endógenas (bilirrubina, ác. Grasos) Efecto farmacológico del fármaco desplazado 3.- En hipoalbuminemias (falla hepática, síndrome nefrótico ) [ F ] libre a cualquier dosis
 Efecto farmacológico. del fármaco desplazado. 3.- En hipoalbuminemias (falla hepática, síndrome nefrótico ) [ F ] libre a cualquier dosis.")
45
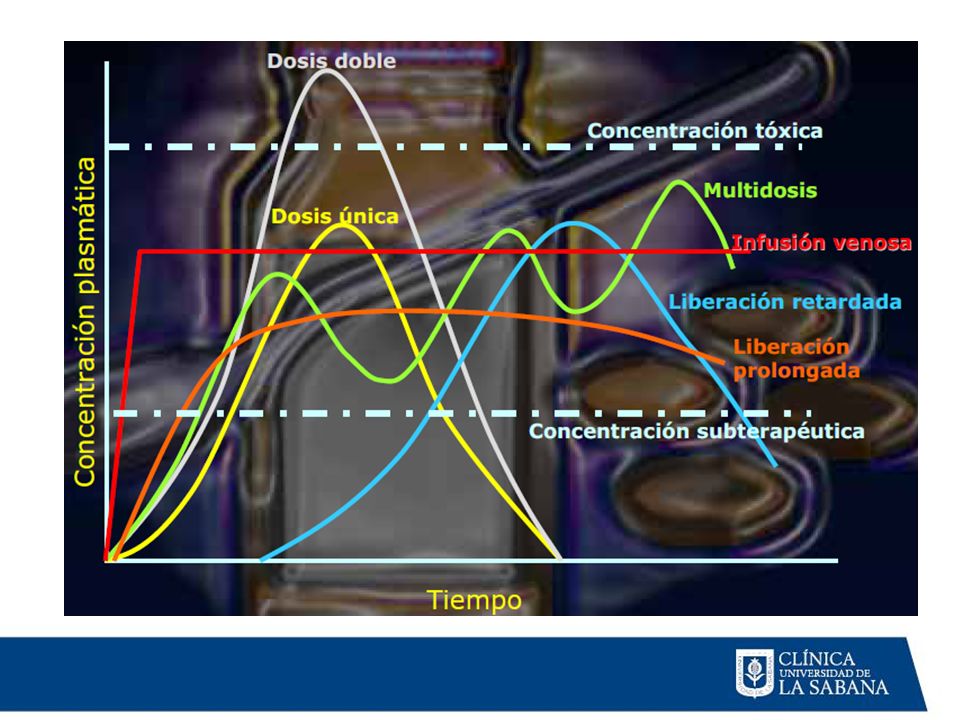
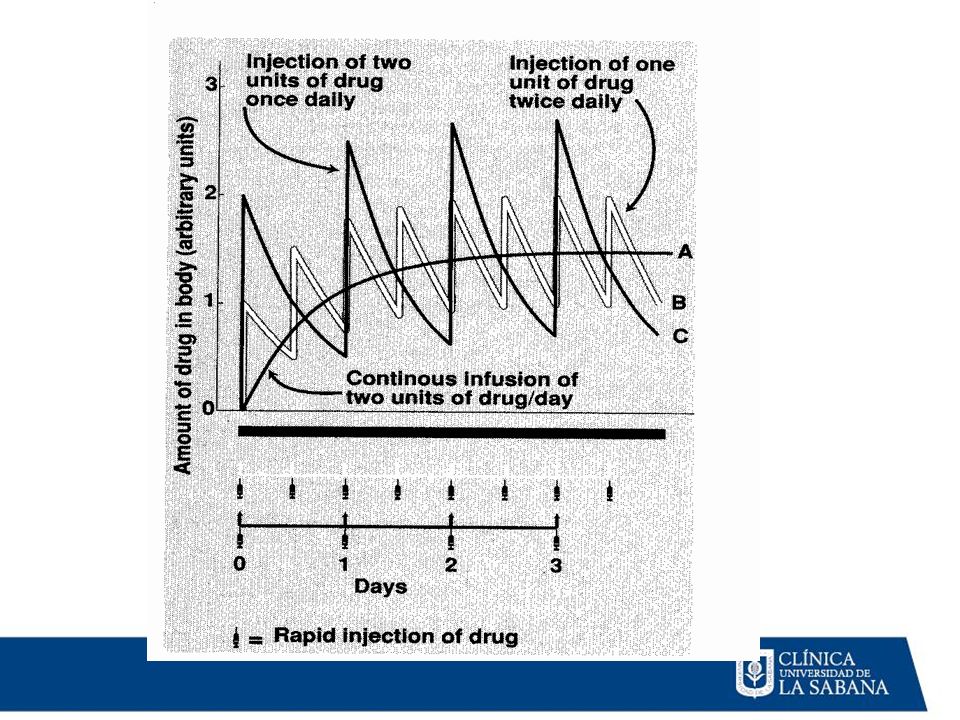
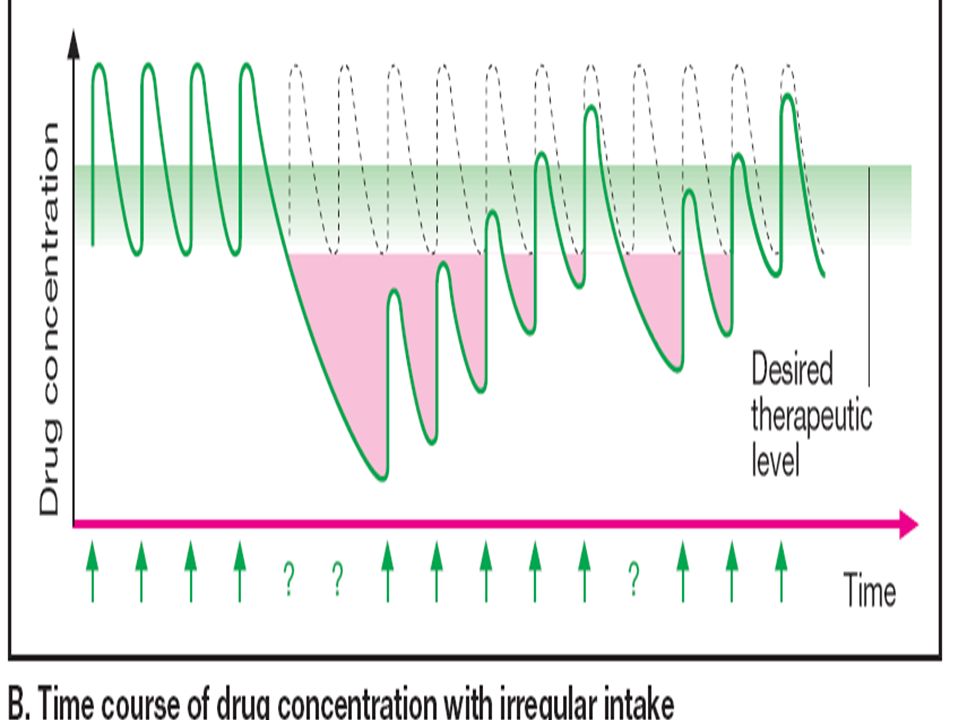
Tiempo (múltiplos de la vida media de eliminación)
Estado de meseta - Se alcanza después de aprox. 5 t1/2 - Tiempo hasta la meseta independientemente de la dosis 2 1 C Concentración Concentraciones de meseta - Proporcionales al intervalo dosis-dosificación - proporcionales a l tiempo medio Fluctuaciones - Proporcionales al intervalo entre dosis/vida media - Amortiguadas por la absorción lenta Tiempo (múltiplos de la vida media de eliminación)
")
46
Tiempo (múltiplos de la vida media de eliminación)
2 1 Concentración Tiempo (múltiplos de la vida media de eliminación)
")
47
Concentración de teofilina (mg/L)
5 10 15 20 25 A B Tiempo (h) Concentración de teofilina (mg/L) A = Infusión intravenosa continua; velocidad de administración de 37.5 mg/h B = Administración en “bolus” intermitente 300 mg cada 8 horas
 Concentración de teofilina (mg/L) A = Infusión intravenosa continua; velocidad de administración de 37.5 mg/h. B = Administración en bolus intermitente 300 mg cada 8 horas.")
50
BIOTRASFORMACION : IMPORTANCIA Favorece la excreción del fármaco, al transformarlo en metabolitos que por lo general son mas polares, mas hidrosolubles menos liposolubles que la molécula madre (esto favorece su excreción y disminuye su Vd) En algunos casos conduce a la inactivación del fármaco o reduce su efecto, al transformarlo en metabolitos inactivos o con menor acción farmacológica que la molécula madre.
 En algunos casos conduce a la inactivación del fármaco o reduce su efecto, al transformarlo en metabolitos inactivos o con menor acción farmacológica que la molécula madre.")
51
En otros casos puede aumentar la acción del fármaco, al transformarlo en metabolitos con mayor efecto que la molécula madre. Esto se aprovecha para administrar un agente inactivo (profármaco) que se convierte en activo dentro del organismo (bioactivación). Los metabolitos pueden ejercer efectos semejantes o diferentes a los de la molécula madre, y pueden ser responsables RAM, toxicidad o interacciones con otros fármacos.

52
MECANISMO DE BIOTRANFORMACION
Se realiza mediante 2 tipos reacciones químicas : Reacciones no sintéticas o de funcionalización (de Fase I): Oxidación, reducción, hidroxilación e hidrólisis. Las reacciones de fase I suelen transformar el fármaco original en un metabolito mas polar, introduciendo o desenmascarando u grupo funcional (-OH,-NH2, -SH). Con frecuencia estos metabolitos son inactivos, aunque en cierto casos solo se modifica su actividad.
: Oxidación, reducción, hidroxilación e hidrólisis. Las reacciones de fase I suelen transformar el fármaco original en un metabolito mas polar, introduciendo o desenmascarando u grupo funcional (-OH,-NH2, -SH). Con frecuencia estos metabolitos son inactivos, aunque en cierto casos solo se modifica su actividad.")
53
Reacciones sintéticas o de biosíntesis (de Fase II):
Conjugación, Acetilación,Metilación y Glutationización : El fármaco o sus metabolitos se acoplan a un compuesto endógeno (ácido glucorónico, sulfato, acetato o un aminoácido). Este proceso habitualmente conduce a la inactivación del fármaco.
. Este proceso habitualmente conduce a la inactivación del fármaco.")
54
Reducción:Cloramfenicol, Clorazepam. Metadona
METABOLISMO o BIOTRANFORMACION REACCIONES DE FASE I Oxidaciones dependientes de citocromo P450:Propranolol, Fenobarbital, Fenitoína, Fenibutazona, Anfetamina, Warfarina. Hidroxilaciones alifáticas: Pentobarbital, Amobarbital, Ibuprofeno, Digitoxina Reducción:Cloramfenicol, Clorazepam. Metadona Hidrólisis:Procaína, Succinilcolina,

55
ConjugaciónReactivo endógeno GlucoronizaciónUDP :ac. Glucorónico
REACCIONES DE FASE II ConjugaciónReactivo endógeno GlucoronizaciónUDP :ac. Glucorónico Morfina, Acetaminofén, Diazepam. Sufatiazol, Digoxina. Acetilación : Acetil-coA Sulfonamidas Isoniazida, Mezcalina. Glutationización: Glutation :Ac. Etacrínico, Bromobenceno Metilación : S-Adenosilmetionina :Dopamina, Adrenalina, Histamina.

56
Farmacocinética: biotransformación, primer paso
Los fármacos que se absorben en el intestino pueden ser biotransformados por enzimas en la pared intestinal y en el hígado antes de llegar a la circulación general Muchos fármacos son convertidos a metabolitos inactivos durante el fenómeno del primer paso, disminuyendo la biodisponibilidad Mabel Valsecia- Farmacologia

57
Metabolismo o Biotransformación

58
Alta hidrosolubilidad
XENOBIOTICO Polares Muy Lipofílicos lábiles estables REACCIONES DE FASE I CONVERSION METABOLICA FASE II CONJUGACIONES ACUMULACION Y SECUESTRO TISULAR (ADIPOCITOS) ELIMINACION RIÑON FILTRACION Productos Polares Alta hidrosolubilidad Productos de ELIMINACION HIGADO BILIS
 ELIMINACION. RIÑON. FILTRACION. Productos Polares. Alta hidrosolubilidad. Productos de. ELIMINACION. HIGADO. BILIS.")
59
BILIAR Y FECAL

60
SIGNIFICADO DE LOS PROCESOS DE
BIOTRANSFORMACION BIOTRANSFORMACION ACTIVACION DESACTIVACION UTILIDAD TERAPEUTICA TOXICIDAD activo inactivo tóxico profármaco Fármaco

61
FACTORES QUE MODIFICAN LA BIOTRANSFORMACIÓN
Factores genéticos: Determinan que el metabolismo de un mismo fármaco pueda ser distinto en individuos de la misma especie. Esto explica la hipersusceptibilidad a ciertos fármacos.

62
FACTORES FISIOLÓGICOS:
Edad. La biotransformación es acelerada en jóvenes y lenta en neonatos (debido a la inmadurez de los sistemas enzimáticos), y en los ancianos (en quienes las funciones renal y hepática se hallan disminuidas). Sexo. Por ej, en la rata hembra el metabolismo de los barbitúricos es más lento que en el macho, debido a que el estradiol (hormona sexual femenina) inhibe la biotransformación de los barbitíricos. Gestación: la placenta constituye un órgano que participa activamente en la biotransformación de fármacos. Estrés: incrementa la biotransformación que inducen la biosíntesis de enzimas metabolizadoras.
, y en los ancianos (en quienes las funciones renal y hepática se hallan disminuidas). Sexo. Por ej, en la rata hembra el metabolismo de los barbitúricos es más lento que en el macho, debido a que el estradiol (hormona sexual femenina) inhibe la biotransformación de los barbitíricos. Gestación: la placenta constituye un órgano que participa activamente en la biotransformación de fármacos. Estrés: incrementa la biotransformación que inducen la biosíntesis de enzimas metabolizadoras.")
63
Los mecanismos de biotransformación están alterados
FACTORES PATOLÓGICOS: Desnutrición Los mecanismos de biotransformación están alterados por falta de los sustratos orgánicos indispensables para tal fin. Insuficiencia hepática: cualquier alteración de la función hepática disminuye la capacidad de este órgano para metabolizar los fármacos. Insuficiencia renal: puede alterar el metabolismo de algunos fármacos que se metabolizan a este nivel.

64
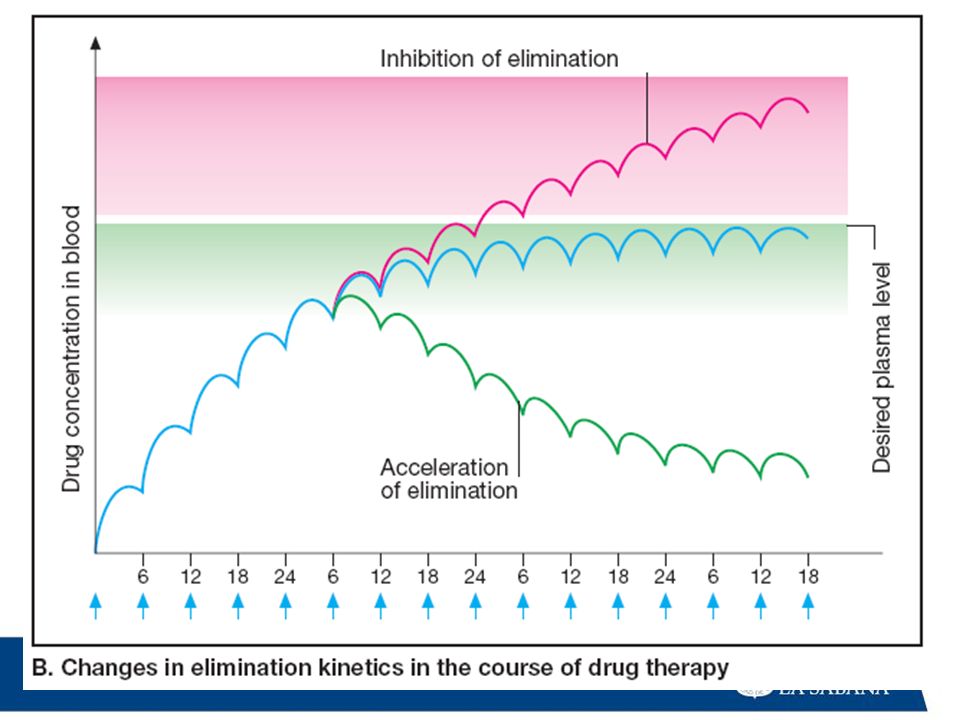
FACTORES FARMACOLÓGICOS:
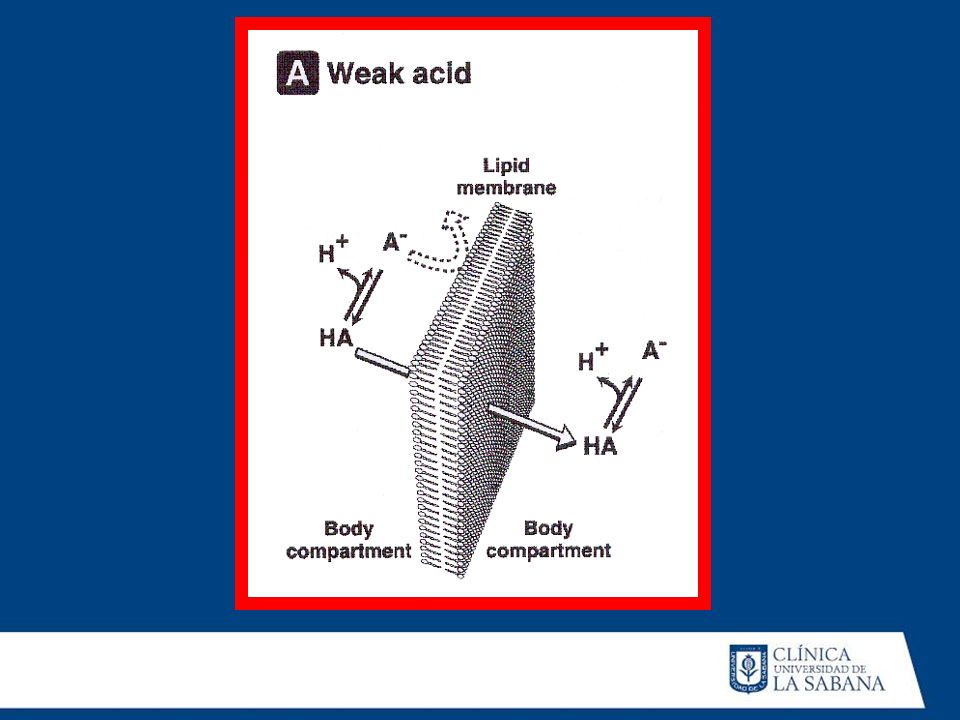
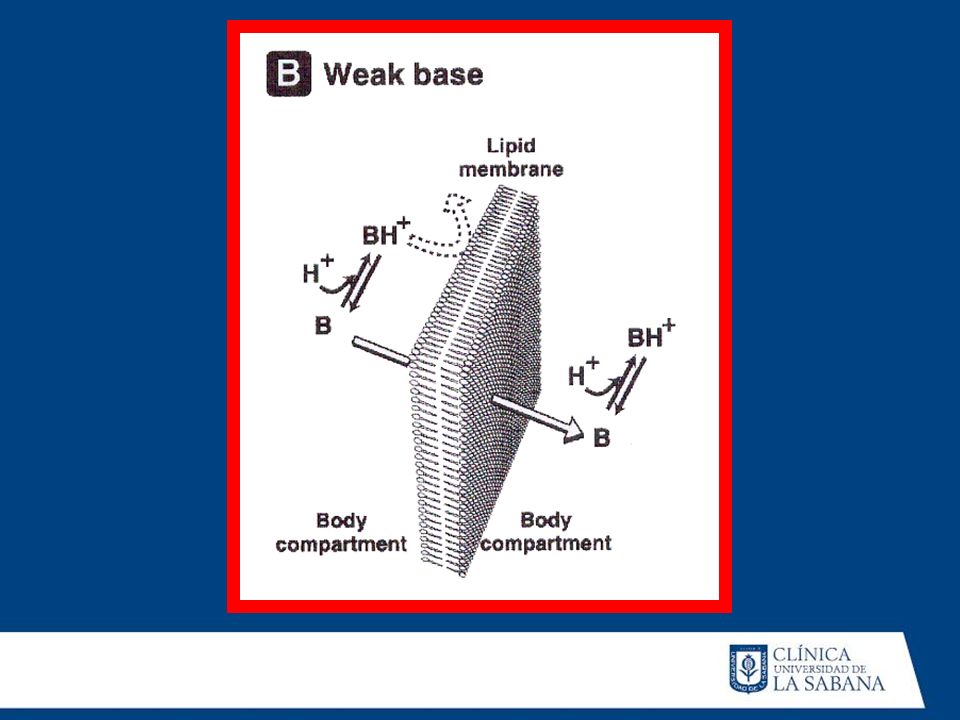
Vía de administración: Los fármacos administrados por VO sufren metabolismo de primer paso, no así los que se administran por vía sublingual o intravascular. Gradientes de pH: Los fármacos existen bajo dos formas (ácidos ybases).Las formas ácidas se excretan en mayor proporción cuando se hallan en medio básico. Si se varía el pH la excreción disminuye y el fármaco tiene mayor posibilidad de metabolizarse.
.Las formas ácidas se excretan en mayor. proporción cuando se hallan en medio básico. Si se. varía el pH la excreción disminuye y el fármaco tiene. mayor posibilidad de metabolizarse.")
65
Inductores enzimáticos:
Algunos fármacos estimulan la síntesis de enzimas microsomales, aumentando su propia biotransformación o la de otros fármacos, lo que origina múltiplres interacciones y efectos tóxicos. Por ej, los babitúricos (considerados entre los más potentes inductores enzimáticos) aceleran su propio metabolismo y el de otros medicamentos. Interacción farmacológica. Los fármacos pueden interactuar entre sí, acelerando o retardando su biotransformación, lo cual puede originar efectos tóxicos. Por ej, la fenilbutazona acelera la biotransformación de los anticoagulantes, inhibiendo su efecto.
 aceleran su propio metabolismo y el de otros medicamentos. Interacción farmacológica. Los fármacos pueden interactuar entre sí, acelerando o. retardando su biotransformación, lo cual puede originar. efectos tóxicos. Por ej, la fenilbutazona acelera la. biotransformación de los anticoagulantes, inhibiendo su efecto.")
66
EXCRECION : EXCRECION : expulsión del fármaco en forma intacta, como ingreso. ELIMINACION: expulsión del fármaco intacto más sus metabolitos procedentes del metabolismo.

67
R I Ñ O N

68
Filtración glomerular:
Todas las sustancias con PM menor a filtran por el glomérulo renal. Los compuestos polares insolubles en agua son incapaces de difundir de regreso a la circulación luego de ser filtrados, por lo que son excretados a menos que exista un sistema de transporte específico para su reabsorción.

70
RIÑON :

71
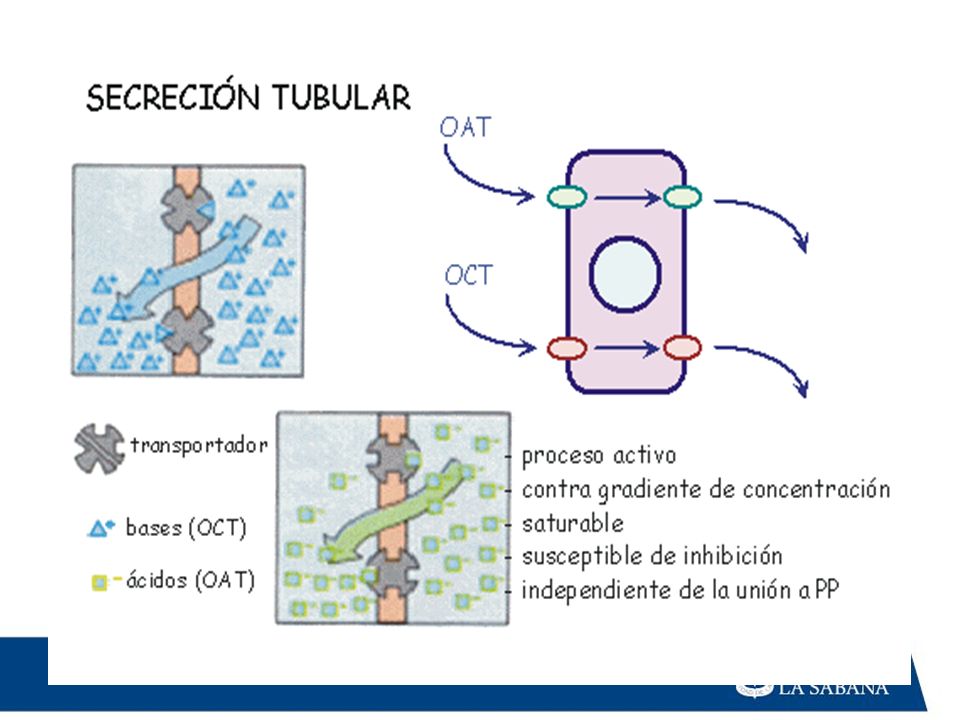
Secreción tubular activa :
Se realiza con los ácidos y bases orgánicas. Los mecanismos de secreción tubular activa se localizan en el túbulo contorneado proximal. Reabsorción tubular pasiva: Ocurre principalmente con los fármacos liposolubles.

72
Excreción urinaria % de la dosis administrada que se elimina en forma intacta (no metabolizada) por el riñón. Ejemplos: % Nifedipina 0 % Warfarina 3 % Acetaminofén Eritromicina 12 % Digoxina 60 % Furosemida 66 % Amikacina 98 % Litio 95 % Ampicilina 82 % Funcion renal / Funcion hepática Clearance renal Clearance hepático Clearance sistémico (Clearance = volumen de plasma limpiado /unidad de tiempo)
")
73
Excreción biliar y fecal:
Muchos metabolismos hepáticos se expulsan con la bilis y pueden eliminarse con las heces, pero lo más común es que sean reabsorbidos del intestino (circulación enterohepática) y se excreten por la orina. Las sustancias excretadas con las heces son principalmente los fármacos ingeridos que no han sido absorbidos, o los metabolitos excretados por la bilis que no fueron reabsorbidos.
 y se excreten por la orina. Las sustancias excretadas con las heces son principalmente los fármacos ingeridos que no han sido absorbidos, o los metabolitos excretados por la bilis que no fueron reabsorbidos.")
74
PULMONAR : Farmacos inhalatorios se eliminan por esta via
La membrana alveolo capilar debe estar indemne Depende del volumen tidal y de las patologías del pulmón Depende del estado neurológico del paciente.

77
CONTESTAR

78
CONTESTAR:

79
FARMACOCINETICA CLINICA

80
FARMACOCINETICA CLINICA
Individualización del régimen de dosificación Detección de interacciones Alteraciones de la biodisponibilidad Resistencia al tratamiento Respuesta ineficaz Intoxicaciones

81
Consideraciones al diseñar la pauta de administración de un fármaco
Características de la enfermedad Eficacia y toxicidad del fármaco Características farmacocinéticas del fármaco y factores que puedan alterarlas. Vía de administración. Forma Farmacéutica que se va a utilizar. Conveniencia de facilitar el cumplimiento terapéutico. Binomio costo/beneficio.

82
Forma Farmacéutica Cápsulas Tabletas Sólidas: Polvos Parches Supositorios Ovulos Comprimidos Grageas

83
Suspensiones Forma Farmacéutica Inyectables Líquidas: Aerosoles Jarabes Soluciones

84
Biodisponibilidad oral
Es el porcentaje de la dosis administrada por vía oral que alcanza la circulación sistémica Se puede expresar como: % de la dosis administrada y fracción biodisponible f MEDICAMENTO FARMACO Factores que condicionan la magnitud de la absorción Metabolismo de primer paso.

86
Dosis de Fármaco Concentración de fármaco libre en el agua
BIOTRANSFORMACIÓN - microsomas hepáticos - no-microsomal - extrahepática Absorción UNIÓN A PROTEINAS PLASMÁTICAS - albúmina Concentración de fármaco libre en el agua extracelular METABOLITOS - inactivos - activos EXCRECIÓN BILIAR - circulación enterohepática UNIÓN Y ALMACENAMIENTO EN TEJIDOS - proteínas - grasa EXCRECIÓN RENAL - filtración glomerular - secreción tubular - reabsorción pasiva Concentración de fármaco en el sitio de acción Patrón de distribución de la cantidad de fármaco en el cuerpo Distribución del fármaco, implica el transporte ( o transferencia) reversible del fármaco de una localización a otra dentro del cuerpo. Una vez que el fármaco ha entrado al sistema vascular este se distribuye a través de varios tejidos y fluidos biológicos en un patrón que refleja la naturaleza fisicoquímica del fármaco y la facilidad con la cual penetra diferentes membranas. Ocupación del receptor Intensidad del efecto farmacológico
 reversible del fármaco de una localización a otra dentro del cuerpo. Una vez que el fármaco ha entrado al sistema vascular este se distribuye a través de varios tejidos y fluidos biológicos en un patrón que refleja la naturaleza fisicoquímica del fármaco y la facilidad con la cual penetra diferentes membranas. Ocupación del receptor. Intensidad del. efecto farmacológico.")
87
Distribución de los fármacos
5% 15% 40% Plasma Agua intersticial Agua intracelular 3- 4 L – 14 L L F. Unido F. unido F. unido Vía i.v F. libre F. libre F. libre Vía oral Hígado Riñón Endotelio capilar Membrana celular Sudor Saliva Leche, etc. Heces Orina Distribución de los fármacos

88
Volumen de distribución.
Volumen en el cual tendría que distribuirse el fármaco para alcanzar en todos los líquidos biológicos una concentración similar a la sanguínea. Vd es un factor de proporcionalidad entre la dosis administrada y la concentración sanguínea. Vd = Dosis i.v Vd = Dosis Cp Cp0 Vd = Dosis administrada (mg) Concentración sanguínea (mg/L)
 . Concentración sanguínea (mg/L)")
89
Ejemplos de volúmenes de distribución de Fármacos
Volumen de distribución (L/Kg) fármaco Plasma 0,05 L/kg 0,05 - 0,1 0,1 - 0,2 Heparina, Insulina Warfarina, Furosemida Líquido extracelular (0,2 L/kg) 0,2 – 0,4 Amikacina, Tubocurarina Agua corporal total (0,55 L/kg) 0, – 5 > 10 Etanol, Fenitoína, Fenobarbital, Diazepam, Indometacina Nitrazepam, Morfina, Digoxina, Imipramina, Nortriptilina
 fármaco. Plasma 0,05 L/kg. 0,05 - 0,1 0,1 - 0,2. Heparina, Insulina Warfarina, Furosemida. Líquido extracelular (0,2 L/kg) 0,2 – 0,4. Amikacina, Tubocurarina. Agua corporal total (0,55 L/kg) 0, – 5 > 10. Etanol, Fenitoína, Fenobarbital, Diazepam, Indometacina Nitrazepam, Morfina, Digoxina, Imipramina, Nortriptilina.")
90
Complejo Fármaco-proteína Fármaco libre Proteína
k k2 Fármaco libre Proteína + Sitio de ACCIÓN ej. pared capilar membrana celular sitio intracelular Sitio de ELIMINACIÓN ej. Filtración glomerular secreción renal tubular biotransformación hepática secreción biliar

92
Importancia clínica de la unión F-P
1.- [ F ] libre actividad farmacológica y clearance renal 2.- Desplazamiento competitivo a.- entre Fármacos b.- por substancias endógenas (bilirrubina, ác. Grasos) Efecto farmacológico del fármaco desplazado 3.- En hipoalbuminemias (falla hepática, síndrome nefrótico ) [ F ] libre a cualquier dosis
 Efecto farmacológico. del fármaco desplazado. 3.- En hipoalbuminemias (falla hepática, síndrome nefrótico ) [ F ] libre a cualquier dosis.")
94
SIGNIFICADO DE LOS PROCESOS DE
BIOTRANSFORMACION BIOTRANSFORMACION ACTIVACION DESACTIVACION UTILIDAD TERAPEUTICA TOXICIDAD activo inactivo tóxico profármaco Fármaco

95
Alta hidrosolubilidad
XENOBIOTICO Polares Muy Lipofílicos lábiles estables REACCIONES DE FASE I CONVERSION METABOLICA FASE II CONJUGACIONES ACUMULACION Y SECUESTRO TISULAR (ADIPOCITOS) ELIMINACION RIÑON FILTRACION Productos Polares Alta hidrosolubilidad Productos de ELIMINACION HIGADO BILIS
 ELIMINACION. RIÑON. FILTRACION. Productos Polares. Alta hidrosolubilidad. Productos de. ELIMINACION. HIGADO. BILIS.")
96
Eliminación de los fármacos:
Riñón Saliva, sudor, leche, lágrimas Pulmones Intactas Bilis Riñón Heces Hígado Riñón Tracto G.I Pulmones Piel Metabolitos Filtración Secreción activa Forma intacta metabolitos Riñón

97
Excreción urinaria % de la dosis administrada que se elimina en forma intacta (no metabolizada) por el riñón. Ejemplos: % Nifedipina 0 % Warfarina 3 % Acetaminofén Eritromicina 12 % Digoxina 60 % Furosemida 66 % Amikacina 98 % Litio 95 % Ampicilina 82 % Funcion renal / Funcion hepática Clearance renal Clearance hepático Clearance sistémico (Clearance = volumen de plasma limpiado /unidad de tiempo)
")
98
Constante de eliminación (Kel)
Fracción o porcentaje del fármaco eliminado por unidad de tiempo (no tiene dimensiones). Se expresa: la fracción por hora o por min. Ejemplo: Kel = 0,15 h-1. Significa que en cada hora se elimina un 15 % de la cantidad del fármaco presente en el cuerpo (o en la sangre que se supone que está en equilibrio en todos los líquidos donde tiene acceso)
. Se expresa: la fracción por hora o por min. Ejemplo: Kel = 0,15 h-1. Significa que en cada hora se elimina un 15 % de la cantidad del fármaco presente en el cuerpo (o en la sangre que se supone que está en equilibrio en todos los líquidos donde tiene acceso)")
99
Se administra por vía i. v
Se administra por vía i.v mg de un fármaco que tiene una Kel= 0,15 h-1 Tiempo (horas) Cantidad de droga en el cuerpo Cantidad eliminada Fracción 1000 - 1 850 150 0.15 2 723 127 3 614 109 4 522 92 5 444 78 6 377 64
 Cantidad de droga. en el cuerpo. Cantidad eliminada. Fracción")
101
Vías de administración y formas farmacéuticas / velocidad de absorción

102
Biodisponibilidad oral
Fármaco % f Ácido Valproico 100 1 Acetaminofén 88 0.88 Ampicilina 35 0.35 Gentamicina Fenobarbital Ciprofloxaciona 95 0.95

103
Cinética y Orden de Reacción
VELOCIDAD fármaco A fármaco B - dA ó dB dt dt Generalmente en forma experimental se mide el fármaco A (farmacológicamente activo) El o los metabolitos normalmente son desconocidos o difíciles de cuantificar
 El o los metabolitos normalmente son desconocidos o difíciles de cuantificar.")
104
CONSTANTE DE VELOCIDAD
El orden de una reacción se refiere a la forma por la cual la concentración del fármaco o reactantes influyen sobre la velocidad de una reacción o proceso químico. Reacciones de Orden Cero Si la cantidad del fármaco A disminuye de manera constante en un intervalo de tiempo t, entonces la velocidad de desaparición del fármaco A es expresado como: (1)
")
105
A m = - k0 A0 t donde k0 es la constante de velocidad de orden cero y se expresa en unidades de masa/tiempo (ej. mg/min) y A0 es la cantidad de fármaco a t = 0
 y A0 es la cantidad de fármaco a t = 0.")
106
Concentración A (mg/mL)
EJEMPLO Concentración A (mg/mL) Tiempo (h) 100 95 2 90 4 85 6 80 8 75 10 70 12 a) Grafique A vs t b) Determine k0
 Tiempo. (h) a) Grafique A vs t. b) Determine k0.")
107
Modelo de 1 compartimiento
Distribución Absorción Kab Eliminación Kel Dosis administrada Fármaco en el cuerpo (velocidad de entrada) (velocidad de salida) Ejemplo más sencillo: bolo intravenoso Dosis I.V. Fármaco en el cuerpo Salida velocidad de salida (Cleareance)
 (velocidad de salida) Ejemplo más sencillo: bolo intravenoso. Dosis I.V. Fármaco en el. cuerpo. Salida. velocidad de salida (Cleareance)")
109
Vía intravenosa dosis única Modelo de 1 compartimiento
100 75 50 25 10 7.5 5.0 2.5 1.0 0.75 0.5 0.25 Concentración del fármaco en plasma (µg/ml) Concentración del fármaco en plasma (µg/ml) Tiempo (h) Tiempo (h) Grafica aritmética Grafica semilogarítmica
 Concentración del fármaco en plasma (µg/ml) Tiempo (h) Tiempo (h) Grafica aritmética. Grafica semilogarítmica.")
110
CONSTANTE DE VELOCIDAD
Reacciones de Primer Orden Si la cantidad del fármaco A disminuye de manera proporcional a la cantidad de fármaco A remanente entonces la velocidad de desaparición del fármaco A se expresa como: (2) donde k es la constante de velocidad de primer orden y se expresa en unidades de tiempo-1 (ej. hr-1)
 donde k es la constante de velocidad de primer orden y se expresa en unidades de tiempo-1 (ej. hr-1)")
111
ln A m = - k A0 t

112
Concentración A (mg/mL)
EJEMPLO Concentración A (mg/mL) Tiempo (h) ln A 100 4,60 50 2 3,91 25 4 3,22 12,5 6 2,53 6,25 8 1,83 3,13 10 1,14 1,56 12 0,44 a) Grafique ln A vs t b) Determine k
 Tiempo. (h) ln A , , ,22. 12, ,53. 6, ,83. 3, ,14. 1, ,44. a) Grafique ln A vs t. b) Determine k.")
113
Modelo de 2 compartimientos
Distribución Fármaco en el compartimiento central Absorción Ka Eliminación Kel Dosis administrada Fármaco en el compartimiento periférico velocidad de entrada velocidad de salida Ejemplo más sencillo: bolo intravenoso (1-2 min): Fármaco en el compartimiento central salida Dosis IV velocidad de salida (Cleareance) Fármaco en el compartimiento periférico
: Fármaco en el compartimiento central. salida. Dosis IV. velocidad de salida (Cleareance) Fármaco en el compartimiento periférico.")
115
Compartimiento periférico Comparti- miento central
32 16 8 4 2 1 C o p Fase de distribución y redistribución T½(a) Concentración plasmática del fármaco (mg/ml) Fase de eliminación T½(b) T1/2 Compartimiento periférico Tiempo (horas) Comparti- miento central
 Concentración plasmática del fármaco (mg/ml) Fase de eliminación. T½(b) T1/ Compartimiento periférico. Tiempo (horas) Comparti- miento central.")
116
Parámetros Farmacocinéticos

117
Relaciones farmacocinéticas fundamentales para la administración repetida de un Fármaco
Vd = 1000 L filtro bomba Vd = 500 L bomba filtro Cl = 25 L/min Cl = 10 L/min 0,7 x Vd Cl t½ = 0,7 x Vd Cl t½ = 0, ,7 0,7 x (1000L) 10L/min 0,7 x (500L) 25L/min = 70 min = 14 min
 10L/min. 0,7 x (500L) 25L/min. = 70 min. = 14 min.")
118
Dosis de Mantenimiento:
Es la cantidad del fármaco que se administra en función de la metabolización y excreción del mismo, permite mantener una concentración plasmática estable (meseta), por tanto se calcula en base al Clearance sistémico del Fármaco. Recuerde que el estado estable (ee) se alcanza 5 t 1/2
, por tanto se calcula en base al Clearance sistémico del Fármaco. Recuerde que el estado estable (ee) se alcanza 5 t 1/2.")
119
Dosis: 300mg Teofilina I.V. (t½ = 6 horas Vd = 30L)
1 2 3 4 5 6 7 8 9 n n + 1 Cantidad O hs (mg) 300 450 525 562.50 581.25 590.63 595.31 596.66 598.83 600 10 15 17.50 18.75 19.38 19.89 19.84 19.92 19.96 20 Cp mg/L Cantidad O hs (mg) 150 225 262 281.25 290.63 295.31 297.66 298.83 299.41 300 Cp mg/L 5.0 7.50 8.75 9.38 9.69 9.84 9.92 9.96 9.98 10 Cantidad eliminada (mg) 150 225 262 281.25 290.63 295.31 297.66 298.83 299.41 300
 Cp mg/L. Cantidad O hs (mg) Cp mg/L Cantidad eliminada (mg)")
120
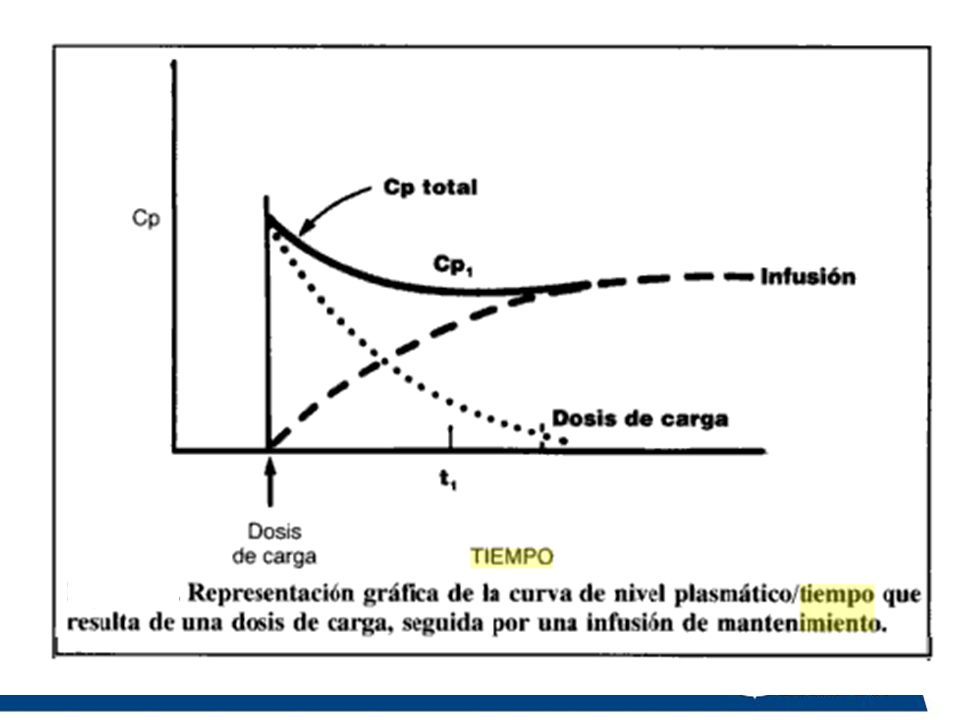
Dosis de carga: Cantidad del fármaco que se administra para alcanzar rápidamente la concentración efectiva, mas no así alcanzar el estado de meseta. Vd = Dosis Vd x Cp deseada = Dosis Cp deseada.

121
Cuando la cantidad que sale (Cl · Cp) iguale a la que entra (Q), la concentración plasmática permanecerá estable tanto tiempo como dure la infusión. Esta concentración denominada en equilibrio (CpE) o nivel estable (Css) y depende directamente de la velocidad de infusión e inversamente del aclaramiento: Donde Q es la velocidad de infusión
, la concentración plasmática permanecerá estable tanto tiempo como dure la infusión. Esta concentración denominada en equilibrio (CpE) o nivel estable (Css) y depende directamente de la velocidad de infusión e inversamente del aclaramiento: Donde Q es la velocidad de infusión.")
122
Velocidad constante de administración (i.v.)
Cp= Vad/Cl Cp= 100 mg/h / (0.2 h-1 * 20 L) Cp= 25 mg/L
 Cp= 25 mg/L.")
123
APLICACION DE CONCEPTOS
Un paciente de 70 Kg de peso debe ser tratado de urgencia con un antibiótico con las siguientes características: Cmax = 80 mg/Lt Cmin = 20 mg/Lt Vd = 3 L/kg t1/2 = 8 h I.V. Esquema de administración Dosis de carga

124
APLICACION DE CONCEPTOS
70 Kg Vd = 3 L/kg t1/2 = 8 h Cmax = 80 mg/Lt Cmin = 20 mg/Lt Esquema de administración t1/2= 0.7/ Ke Ke= 0.7/t1/2 Ke= 0.7/8 h Ke= h-1 Cp= Vad/Cl Vad= Cp * Cl Vad= 50 mg/L * Ke*Vd Vad= 50 mg/L * h-1 * 3L/kg Vad= 50 mg/L * L/h Kg Vad= 13.1 mg/kg /h Vad= 918 mg/h Vad= 22 g en 24 h en infusión continua Dosis= 2.8 g c/8h Dosis de Carga: Cmax*Vd: 80 mg/L*3L/Kg*70 Kg= 16.8 g

125
Papel de la Farmacocinética Clínica

128
Monitorización según el Paciente

130
GRACIAS www.evidenciaterapeutica.com juliogc@clinicaunisabana.edu.co

Presentaciones similares