Descargar la presentación
La descarga está en progreso. Por favor, espere

1
DRA MARIA ELENA SOTO SINDROME DE MARFAN.

2
Introduccion El Síndrome de Marfan (SMF) es un una enfermedad autosómica dominante con una considerable variabilidad intra e interfamiliar, no hay una predisposición por etnia o por genero.
 es un una enfermedad autosómica dominante con una considerable variabilidad intra e interfamiliar, no hay una predisposición por etnia o por genero.")
3
MARFAN Antoine Marfan-Bernard pediatra francés en 1896 describió el primer caso de deformidad congénita con alargamiento de las cuatro extremidades, propuso llamarlo dolichosténomélie; en una niña de 5 años de edad de nombre Gabrielle, con manifestaciones óseas de la enfermedad que hoy lleva su nombre, se ha tenido un importante avance en la delimitación del SMF y el reconocimiento de riesgos asociados

4
EPIDEMIOLOGIA Incidencia 1/10,000 habitantes
Los antecedentes familiares de SM se encuentran hasta en 49% de las familias de personas con el padecimiento. En 25º 30% de los pacientes la enfermedad ocurre sin historia familiar positiva.

5
ETIOLOGIA Mutación autosómica dominante del gen FBN1

7
Los criterios diagnósticos de Ghent son más sensibles y específicos que los criterios de Berlín, el sub-diagnóstico de SMF y la optimización para una mejor diferenciar de SMF relacionados.

8
Criterios Mayores Ocular Cardiovascular Familiar Genomico
Musculoesqueletico

9
Diagnostico

10
Puntuación de alteraciones musculo esqueléticas. SCORESISTEMICO
Características clínica SCORE Signo de la muñeca (signo de Steimberg) y el pulgar (signo de Walker-Murdoch) Signo de la muñeca o el pulgar 3 1 Deformidad del tórax (Pectum carinatum) Pectus excavatum o asimetría del tórax 2 Pie cavo Pie normal o plano Neumotórax Ectasia dural Protrusión acetabular Reducción de US / LS* y un incremento en la relación brazo/talla y con escoliosis no severa Escoliosis o la xifosis toracolumbar Reducción en la extensión del codo Rasgos faciales (3/5) (dolicocefalia, enoftalmos, Inclinación baja de las comisuras palpebrales, hipoplasia malar, retrognatia) Piel con estrías Miopía> 3 dioptrías Prolapso de la válvula mitral (todo tipo) Total: 20 puntos, puntuación > 7 indica participación sistémica. *US/ LS, segmento superior/segmento inferior 20 Los criterios para el diagnóstico de SMF (Criterios de Ghent) se basan principalmente en los hallazgos clínicos en los diversos sistemas de órganos, esbozan la opinión de expertos internacionales; que facilitan el reconocimiento de este síndrome y favorecen el asesoramiento y manejo de los pacientes
 y el pulgar (signo de Walker-Murdoch) Signo de la muñeca o el pulgar Deformidad del tórax (Pectum carinatum) Pectus excavatum o asimetría del tórax. 2. Pie cavo. Pie normal o plano. Neumotórax. Ectasia dural. Protrusión acetabular. Reducción de US / LS* y un incremento en la relación brazo/talla y con escoliosis no severa. Escoliosis o la xifosis toracolumbar. Reducción en la extensión del codo. Rasgos faciales (3/5) (dolicocefalia, enoftalmos, Inclinación baja de las comisuras palpebrales, hipoplasia malar, retrognatia) Piel con estrías. Miopía> 3 dioptrías. Prolapso de la válvula mitral (todo tipo) Total: 20 puntos, puntuación > 7 indica participación sistémica. *US/ LS, segmento superior/segmento inferior. 20. Los criterios para el diagnóstico de SMF (Criterios de Ghent) se basan principalmente en los hallazgos clínicos en los diversos sistemas de órganos, esbozan la opinión de expertos internacionales; que facilitan el reconocimiento de este síndrome y favorecen el asesoramiento y manejo de los pacientes.")
11
DIAGNOSTICO Si no hay antecedentes familiares de SM, entonces el individuo tiene la enfermedad en cualquiera de las siguientes cuatro situaciones: - Dilatación de la raíz aórtica y subluxación de cristalino o - Dilatación de la raíz aórtica y una mutación en FBN1 que es claramente patológica o - Dilatación de la raíz aórtica y características sistémicas múltiples o - Subluxación de cristalino y una mutación en FBN1 que se ha asociado previamente con enfermedad aórtica

12
DIAGNOSTICO Si hay un antecedente familiar positivo de SM
- Subluxación de cristalino o - Características sistémicas múltiples o - Dilatación de la raíz aórtica

13
En la ausencia de historia familiar
1 Diámetro de la raíz aortica (Z score >2 y Ectopia lentis = SM 2 Diámetro de la raíz aortica (Z score > 2 y una mutación de FBN-1 3 Diámetro de la raíz aórtica (Z score >2 y un score sistémico > 7 puntos 4 Ectopia Lentis y una mutación causal conocida de dilatación de la aorta En la presencia de historia familiar 5 Ectopia lentisy historia familiar de SM 6 Score sistémico > 7 puntos y historia familiar del SM 7 Diámetro de la raíz aortica Z score >2 en sujetos mayores a 20 años y > 3 en menores de 20 años. Considerando características diferenciales; Shprintzen Goldberg, Síndrome de Loeys-Dietz, síndrome de Ehlers Danlos o la forma vascular del mismo, otras características bioquímicas de TGBFR1 y TGBR2, o COL3A1 están indicadas.

14
Diagnóstico diferencial
Gen Características clínicas Síndrome de Loeys-Dietz (SLD) TGFBR1/2 Úvula bífida, paladar hendido, tortuosidad arterial, hipertelorismo, aneurisma aórtico y arterial, craneosisnostosis, pie varo, inestabilidad cervical, piel fina y aterciopelada, fragilidad capilar (equimosis) Síndrome de Sphrintzen- Goldberg (SGS) FBN1 y otros Craneosinostosis, retardo metal. Aracnodactilia Congénita Contracturada (CCA) FBN2 Contracturas, pabellones auriculares arrugados. Síndrome de Weill-Marchesani (WMS) FBN1 y ADAMTS10 Microesferofaquia, braquidactilia, rigidez en las articulaciones. Síndrome de Ectopia Lentis (ELS) FBN1,LTBP2,ADAMTSL4 Ausencia de dilatación de raíz aórtica Homocistenuria CBS Trombosis, retraso mental. Síndrome de aneurisma torácico familiar (FTAA) FTAA con aorta bivalva (BAV) FTAA con persistencia de conducto arterioso (PDA) TGFBR1/2,ACTA2 MYH11 Sin características esqueléticas con síndrome de Marfan, livedo reticularis, floculación del iris. Síndrome de tortuosidad arterial (ATS) SLC2A10 Tortuosidad arterial generalizada, estenosis arterial, dismorfismo arterial. Síndrome de Ehelers-Danlos (Tipo vascular, valvular y con xifoescoliosis) COL3A1, COL1A2, PLOD1 Aneurismas arteriales, insuficiencia valvular severa, cicatrices dismorficas, piel traslucida. Síndrome Beals-Heacht Crom 5 (5q23-31). Manifestación al nacimiento, artrogriposis, contracturas musculares congénitas, aracnodactilia, dolicostenomegalia, xifoescoliosis, malformación en las orejas (orejas de sopillo) Síndrome de Marshall Crom 12 (12q14) Sordera, miopía, catarata y nariz en silla de montar. Síndrome de Camurati-Engelman TGFB1 Cansancio fácil; Marcha tambaleante; Dolor en las piernas; Hipotrofia muscular; piernas arqueadas; rodillas en valgo; pies planos y pronados; reflejos tendinosos exaltados y clonus aquíleo; afectación de pares craneales por esclerosis Síndrome de hipermobilidad articular COL1, COL5 Dolores articulares, bursitis, tendinitis, subluxaciones articulares, dolor de espalda, etc.) como de otros tejidos: prolapso uterino o rectal, hernias abdominales, venas varicosas, piel delgada (transparente) con estrías, fragilidad capilar y mala cicatrización, prolapso de la válvula mitral, miopía, párpados caídos, Síndrome de Furlong Así era llamado el Sx Loetz-Dietz Síndrome de Marfan Neonatal FBN1 Ecocardiografía prenatal se ha detectado cardiomegalia con insuficiencia tricúspidea severa. Al nacer, se evidencia alteraciones esqueléticas y de piel (extremidades largas con dedos finos, aspecto envejecido, piel laxa, hipotonía, alteraciones del tórax, contracturas en flexión, micrognatia) cardiovasculares (insuficiencia mitral y tricúspidea severas, cardiomegalia, dilatación aórtica y pulmonar, arritmias, prolapso mitral y tricúspideo, aneurismas masivos de la aorta ascendente y descendente). La muerte ocurre en horas o días por insuficiencia cardíaca
 TGFBR1/2. Úvula bífida, paladar hendido, tortuosidad arterial, hipertelorismo, aneurisma aórtico y arterial, craneosisnostosis, pie varo, inestabilidad cervical, piel fina y aterciopelada, fragilidad capilar (equimosis) Síndrome de Sphrintzen- Goldberg (SGS) FBN1 y otros. Craneosinostosis, retardo metal. Aracnodactilia Congénita Contracturada (CCA) FBN2. Contracturas, pabellones auriculares arrugados. Síndrome de Weill-Marchesani (WMS) FBN1 y ADAMTS10. Microesferofaquia, braquidactilia, rigidez en las articulaciones. Síndrome de Ectopia Lentis (ELS) FBN1,LTBP2,ADAMTSL4. Ausencia de dilatación de raíz aórtica. Homocistenuria. CBS. Trombosis, retraso mental. Síndrome de aneurisma torácico familiar (FTAA) FTAA con aorta bivalva (BAV) FTAA con persistencia de conducto arterioso (PDA) TGFBR1/2,ACTA2. MYH11. Sin características esqueléticas con síndrome de Marfan, livedo reticularis, floculación del iris. Síndrome de tortuosidad arterial (ATS) SLC2A10. Tortuosidad arterial generalizada, estenosis arterial, dismorfismo arterial. Síndrome de Ehelers-Danlos (Tipo vascular, valvular y con xifoescoliosis) COL3A1, COL1A2, PLOD1. Aneurismas arteriales, insuficiencia valvular severa, cicatrices dismorficas, piel traslucida. Síndrome Beals-Heacht. Crom 5 (5q23-31). Manifestación al nacimiento, artrogriposis, contracturas musculares congénitas, aracnodactilia, dolicostenomegalia, xifoescoliosis, malformación en las orejas (orejas de sopillo) Síndrome de Marshall. Crom 12 (12q14) Sordera, miopía, catarata y nariz en silla de montar. Síndrome de Camurati-Engelman. TGFB1. Cansancio fácil; Marcha tambaleante; Dolor en las piernas; Hipotrofia muscular; piernas arqueadas; rodillas en valgo; pies planos y pronados; reflejos tendinosos exaltados y clonus aquíleo; afectación de pares craneales por esclerosis. Síndrome de hipermobilidad articular. COL1, COL5. Dolores articulares, bursitis, tendinitis, subluxaciones articulares, dolor de espalda, etc.) como de otros tejidos: prolapso uterino o rectal, hernias abdominales, venas varicosas, piel delgada (transparente) con estrías, fragilidad capilar y mala cicatrización, prolapso de la válvula mitral, miopía, párpados caídos, Síndrome de Furlong. Así era llamado el Sx Loetz-Dietz. Síndrome de Marfan Neonatal. FBN1. Ecocardiografía prenatal se ha detectado cardiomegalia con insuficiencia tricúspidea severa. Al nacer, se evidencia alteraciones esqueléticas y de piel (extremidades largas con dedos finos, aspecto envejecido, piel laxa, hipotonía, alteraciones del tórax, contracturas en flexión, micrognatia) cardiovasculares (insuficiencia mitral y tricúspidea severas, cardiomegalia, dilatación aórtica y pulmonar, arritmias, prolapso mitral y tricúspideo, aneurismas masivos de la aorta ascendente y descendente). La muerte ocurre en horas o días por insuficiencia cardíaca.")
15
CRITERIO FAMILIAR.

16
Criterio ocular

17
CRITERIO CARDIOVASCULAR

18
CRITERIO SCORE SISTEMICO
Características clínica SCORE Signo de la muñeca (signo de Steimberg) y el pulgar (signo de Walker-Murdoch) Signo de la muñeca o el pulgar 3 1 Deformidad del tórax (Pectum carinatum) Pectus excavatum o asimetría del tórax 2 Pie cavo Pie normal o plano Neumotórax Ectasia dural Protrusión acetabular Reducción de US / LS* y un incremento en la relación brazo/talla y con escoliosis no severa Escoliosis o la xifosis toracolumbar Reducción en la extensión del codo Rasgos faciales (3/5) (dolicocefalia, enoftalmos, Inclinación baja de las comisuras palpebrales, hipoplasia malar, retrognatia) Piel con estrías Miopía> 3 dioptrías Prolapso de la válvula mitral (todo tipo) Total: 20 puntos, puntuación > 7 indica participación sistémica. *US/ LS, segmento superior/segmento inferior 20
 y el pulgar (signo de Walker-Murdoch) Signo de la muñeca o el pulgar Deformidad del tórax (Pectum carinatum) Pectus excavatum o asimetría del tórax. 2. Pie cavo. Pie normal o plano. Neumotórax. Ectasia dural. Protrusión acetabular. Reducción de US / LS* y un incremento en la relación brazo/talla y con escoliosis no severa. Escoliosis o la xifosis toracolumbar. Reducción en la extensión del codo. Rasgos faciales (3/5) (dolicocefalia, enoftalmos, Inclinación baja de las comisuras palpebrales, hipoplasia malar, retrognatia) Piel con estrías. Miopía> 3 dioptrías. Prolapso de la válvula mitral (todo tipo) Total: 20 puntos, puntuación > 7 indica participación sistémica. *US/ LS, segmento superior/segmento inferior. 20.")
19
Steinberg – Walker Murdock

23
Deformidad de torax.

24
DIFERENCIAS de LOEYS DIETZ Vs Marfan
Arterias tortuosas Aneurismas frecuentes y disección en otros sitios de la aorta o en otras arterias distintas de la aorta Hipertelorismo Úvula grande, ancha, bífida Paladar hendido Pie equino varo

25
DIFERENCIAS Escleras azules
Lesiones fáciles, cicatrices anormales y aspecto translúcido que permite ver las venas debajo de la piel sin dificultad Defectos cardiacos al nacimiento, como defecto del septo auricular, persistencia del conducto arterioso y válvula aórtica bicúspide.

26
DIFERENCIAS Craneosinostosis Hidrocefalia
Malformación o inestabilidad de la columna cervical Órganos frágiles que puede provocar ruptura del bazo y de la vejiga o del útero durante el embarazo

29
Caracteristicas clínicas en Síndrome de Marfan y padecimientos similares

30
Síndrome de Marfan

31
Loeys Dietz

32
Experimental Dermatology, 17, 362–365

33
Shprintzen Goldberg

34
Camurati-Engelman Cromosoma 19q Clin Genet 2001: 59: 198–200

35
Weill Marchesanni

36
Neonatal Síndrome de Beals-Hecht Hecht.

37
Aracnodactilia contractural congénita.
Hallazgo CCA VDEGS Paciente CRANEOFACIAL Blefarofimosis Iris coloboma Puente nasal chato Hipoplasia malar Orejas prominentes Orejas deformes nariz picuda Labios evertidos Paladar ojival Retrognatia ESQUELETICO Aracnodactilia Campodactilia Dedos alargados y doblados Contracturas articulares Metacarpos y falanges largos y delgados - +

Presentaciones similares

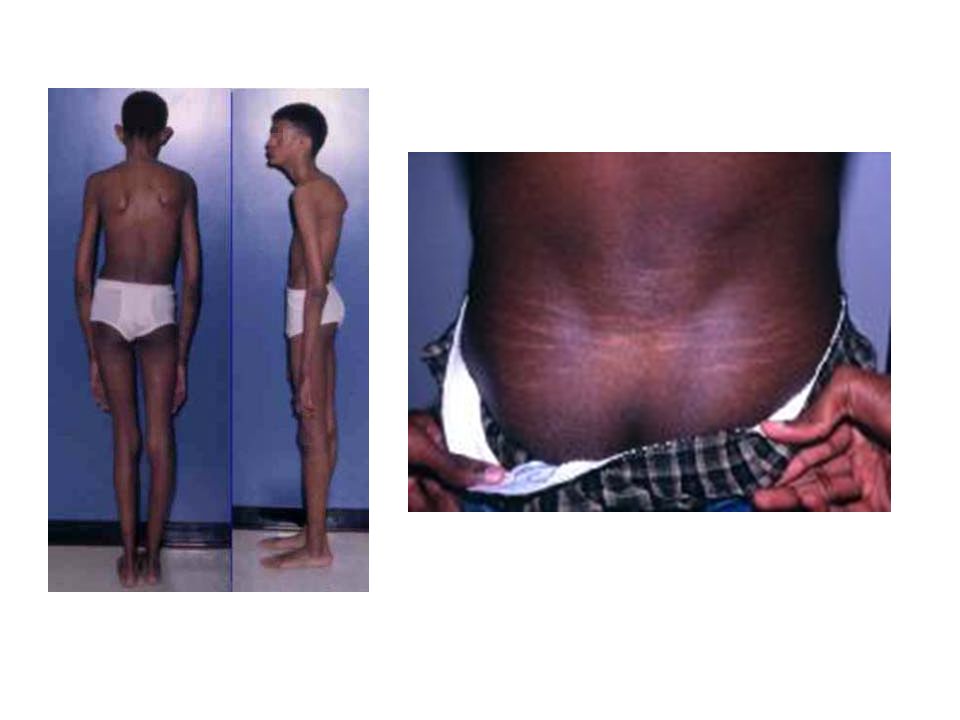
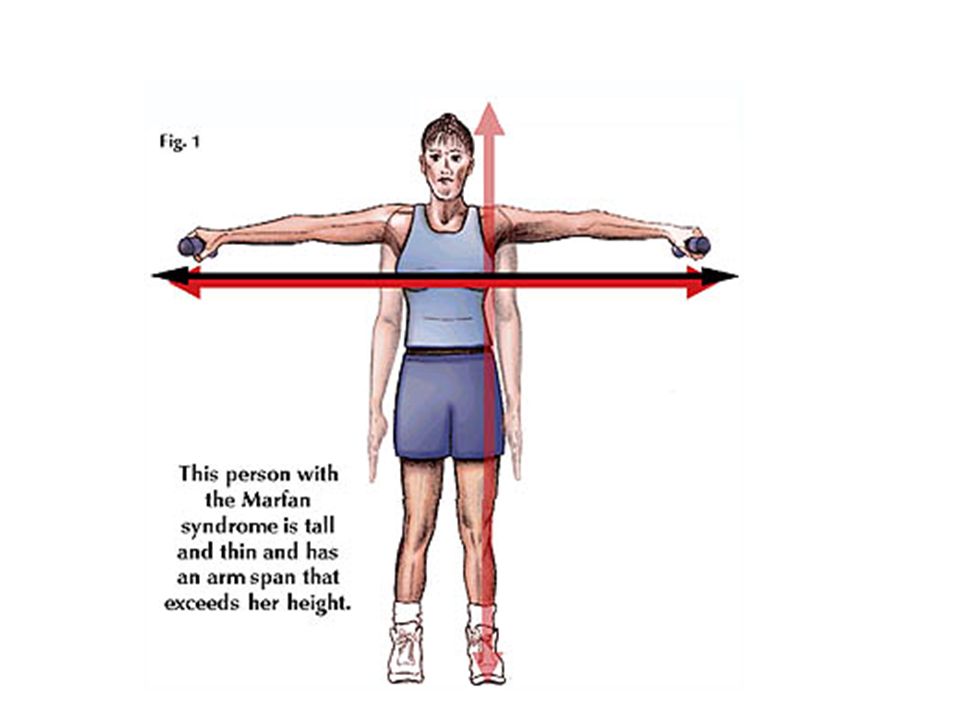
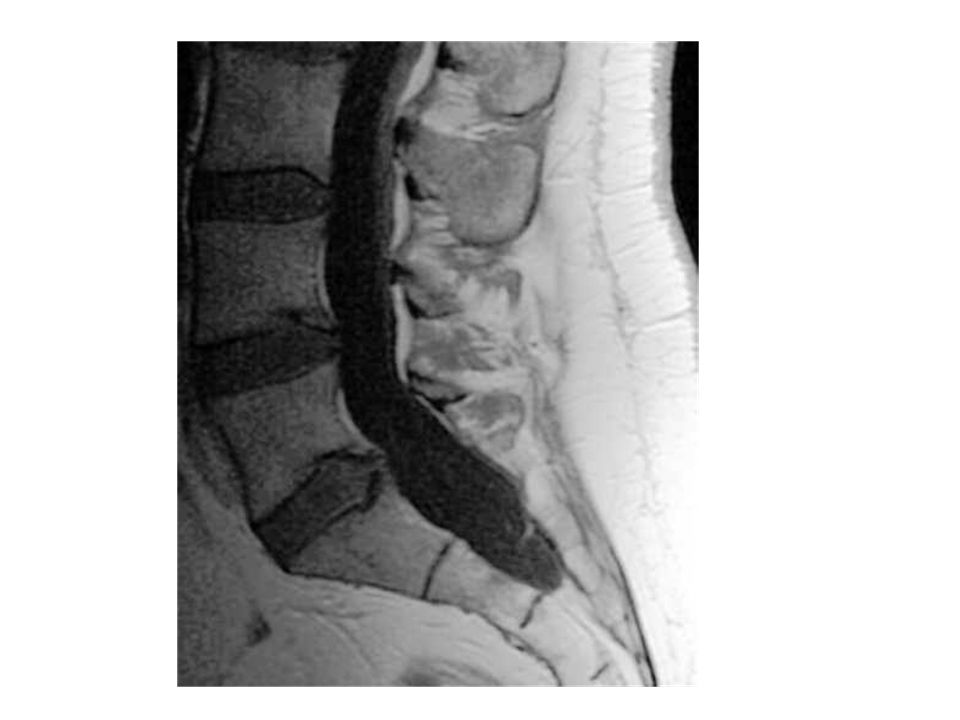
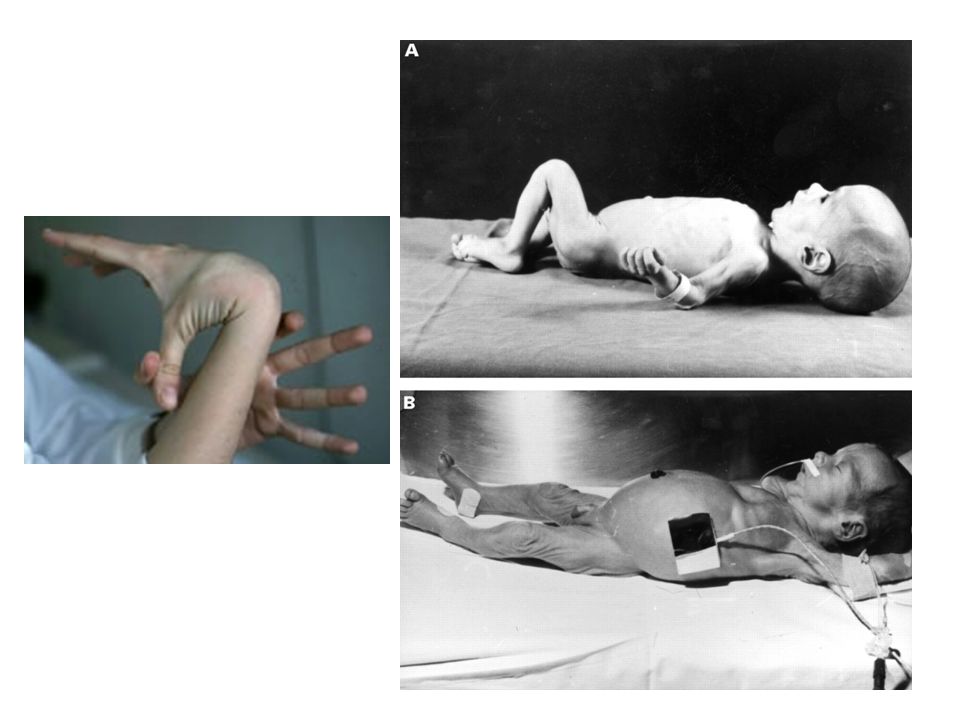


















, es decir, del corazón y de los vasos sanguíneos, son:>")





