Descargar la presentación
La descarga está en progreso. Por favor, espere

1
Dra. Leticia Yáñez P. UPCP Clínica Santa María Rama Intensivo Pediátrico SOCHIPE CETOACIDOSIS DIABÉTICA EN NIÑOS

3
TEMAS Definición ketoacidosis diabética Epidemiología KAD Fisiología control de glicemia Patogénesis KAD Características clínicas y diagnóstico KAD Tratamiento KAD Complicaciones Resumen

4
DEFINICIONES

5
Cetoacidosis Diabética HIPERGLICEMIA glucosa sanguínea > 200 mg/dl (11 mmol/L) ACIDOSIS METABÓLICA pH < 7,3 ó HCO3 plasmático < 15 mEq/L (15 mmol/L) Leve7,2 – 7,3 Moderada7,1 – 7,3 Severa< 7,1 ELEVACIÓN DE CUERPOS CETÓNICOS > 5 mmol/L OSMOLARIDADVariable
 ACIDOSIS METABÓLICA pH < 7,3 ó HCO3 plasmático < 15 mEq/L (15 mmol/L) Leve7,2 – 7,3 Moderada7,1 – 7,3 Severa< 7,1 ELEVACIÓN DE CUERPOS CETÓNICOS > 5 mmol/L OSMOLARIDADVariable")
6
Estado Hiperglicémico Hiperosmolar HIPERGLICEMIAGlicemia >600 mg/dl) ACIDOSIS METABÓLICA pH > 7,3 ó HCO3 plasmático > 18 mEq/L (18 mmol/L) CUERPOS CETÓNICOSNormales o levemente elevados OSMOLARIDAD> 320 mOsm/L Adolescentes afroamericanos con DM tipo II Se asocia a mayor deshidratación y dificultad de manejo de hipotensión Adolescentes afroamericanos con DM tipo II Se asocia a mayor deshidratación y dificultad de manejo de hipotensión
 ACIDOSIS METABÓLICA pH > 7,3 ó HCO3 plasmático > 18 mEq/L (18 mmol/L) CUERPOS CETÓNICOSNormales o levemente elevados OSMOLARIDAD> 320 mOsm/L Adolescentes afroamericanos con DM tipo II Se asocia a mayor deshidratación y dificultad de manejo de hipotensión Adolescentes afroamericanos con DM tipo II Se asocia a mayor deshidratación y dificultad de manejo de hipotensión")
7
EPIDEMIOLOGÍA

8
Epidemiología CAD DM tipo I Debut generalmente –15 a 70 % –Inglaterra 38% EEUU 25% Alemania 26% Principal causa de morbilidad, mortalidad y hospitalización en DM I establecida Mortalidad: –0,15 – 0,5% –Edema cerebral 60 – 90%

9
Epidemiología CAD DM tipo II: –En condiciones de stress –< frecuente en debut –13% KAD tienen DM tipo II –Incidencia ha aumentado en algunos grupos étnicos Canadá 4% Irlanda 25% México 30 % En adolescentes obesos afro- Americanos 40%

10
CAD DM Tipo I Presentación inicial –Mayor riesgo en < 5 años –Nivel socioeconómico inferior –Diagnóstico tardío –Niños de países con baja prevalencia de DM I

11
CAD DM Tipo I Presentación en DM I establecida Factores precipitantes Falta de insulina Stress Drogas: corticoides, antipsicoticos atipicos, tiazidas, diasoxido –Factores de riesgo –Altos requerimientos de insulina –Gastroenteritis (vómitos, deshidratación) –Mujeres adolescentes (> riesgo en > 13 años) –Alteraciones psiquiátricas o disfunción familiar –Falta de previsión de salud –Omisión inadvertida de insulina
 –Mujeres adolescentes (> riesgo en > 13 años) –Alteraciones psiquiátricas o disfunción familiar –Falta de previsión de salud –Omisión inadvertida de insulina")
12
FISIOLOGÍA CONTROL GLICEMIA

13
Ingesta de glucosa Inhibición de liberación de glucagón

14
Fisiología: Glucagón

15
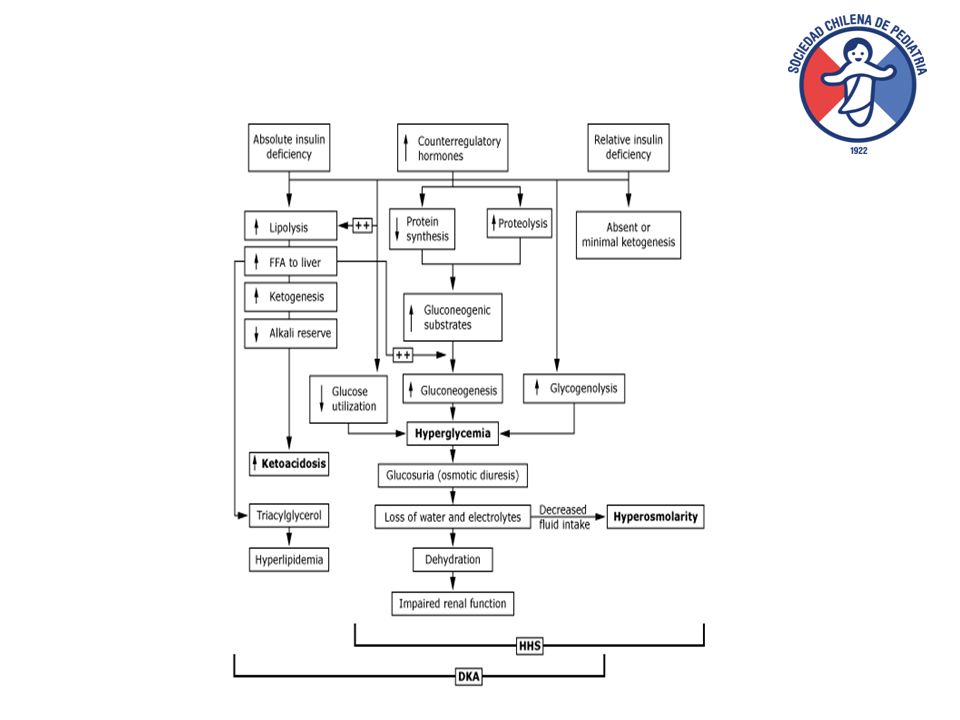
Patogénesis CAD

16
Deficiencia de Insulina y/o resistencia 1 Exceso de Glucagón, no es requisito 2 Aumento secreción de catecolaminas y cortisol 3 Patogénesis Hiperglicemia

18
Deficiencia de Insulina 1 Exceso de Glucagón 2 Se cr ec ió n de ca te co la mi na s y co rti so l 3 Patogénesis Ketoacidosis

19
Lipolisis, que aumenta entrega de ácidos grasos al hígad Ácil CoA entra a la mitocondria y se transforma en ketonas Entrada a la mitocondria es regulada por enzima Carnitinpalmitoiltransferasa (CPT). Glucagón aumenta actividad de CPT y por lo tanto aumenta ketogénesis

21
Resumen KAD > frecuente en DM tipo I KAD – EHH consecuencias del déficit de insulina y exceso de glucagón Hiperglicemia: alteración en utilización de glucosa, aumento de glicogenolisis, neoglucogenesis, lipolisis Niveles de hiperglicemia EHH>KAD Ketoacidosis es << frecuente en EHH

22
Características clínicas Diagnóstico

23
Diagnóstico Alto índice de sospecha Síntomas relacionados con –Hiperglicemia –Acidosis –Hipovolemia –Hiperosmolaridad

24
Diagnóstico: Hiperglicemia Síntomas precoces en niños mayores y adolescentes –Poliuria –Polidipsia –Fatiga –Pérdida de peso –Nicturia – Enuresis nocturna 2ª –Moniliasis vaginal o cutánea –Polifagia (precoz)
")
25
Diagnóstico: Acidosis Vómitos, dolor abdominal Aliento cetónico Hiperventilación profunda, respiración de Kussmaul Aumento de ventilación minuto

26
Diagnóstico: Hipovolemia Difícil diagnóstico Densidad urinaria no sirve (glucosa, ketonas) Mucosas secas, disminución de turgor de piel menos evidente Reflejo de pérdida de agua > sodio –Diuresis osmotica –Hiperventilación –Distribución libre del agua en intra y extracelular
 Mucosas secas, disminución de turgor de piel menos evidente Reflejo de pérdida de agua > sodio –Diuresis osmotica –Hiperventilación –Distribución libre del agua en intra y extracelular")
27
Diagnóstico: Hiperosmolaridad - acidosis Alteraciones neurológicas Decaimiento Letargia Obnubilación Coma

28
Déficit de fluidos y electrolitos Pérdidas estimadas –Agua70 (30 – 100) ml/kg –Sodio5 – 13 mEq/kg –Potasio6 – 7 mEq/kg Difícil evaluación de pérdidas –5 – 10% del peso
 ml/kg –Sodio5 – 13 mEq/kg –Potasio6 – 7 mEq/kg Difícil evaluación de pérdidas –5 – 10% del peso")
29
Hallazgos de laboratorio Hiperglicemia > 200 mg% Gases: Bicarbonato < 15 mEq/L, pH<7,3 La severidad de la acidosis dependerá de Tasa de producción de cuerpos cetónicos Duración de la producción aumentada de cc Tasa de excreción urinaria de ácidos Acido acetico B hidroxibutirato (sangre, > 3 mmol/L o 31 mg/dl) Cetonas
 Cetonas")
30
Laboratorio Índice de –Severidad de ketosis –Resolución de la ketonemia Si está puede reflejar hipoperfusión tisular y renal ANION GAP Na – (Cl + HCO3) Valor normal: 12 ± 2 mmol/L = mEq/L La pérdida de cc en orina (sales de BOHB y < de AA de sodio o potasio) disminuye AG sin alterar HCO3 plasm y el grado de acidosis. Permite monitorizacion del tratamiento

31
Laboratorio: Sodio Natremia Estudios en adultos : –Natremia disminuye 2,4 meq/L por cada 100 mg% de elevación de glicemia MECANISMOS Hiperglicemia aumenta osmolaridad, arrastra agua desde el intracelular y genera hiponatremia Glucosuria induce diuresis osmótica, con pérdida urinaria de Na, K agua y osmolaridad Hiperlipidemia produce pseudohiponatremia

32
Laboratorio: Potasio Pérdidas de K por: Diuresis osmótica Excreción de ketoácidos Pérdidas gastrointestinales Hiperaldosteronismo 2º Déficit de insulina impide entrada de K a célula Hiperosmolaridad saca agua y K desde la célula Tendencia a elevar calemia En promedio en niños: 6 – 7 mEq/kg

33
Laboratorio: Fósforo Disminución de la ingesta Pérdidas de P por: –Diuresis osmótica HIPOFOSFEMIA ENMASCARADA Favorecen salida de la célula: Déficit de insulina Acidosis metabólica

34
Laboratorio: Nitrógeno ureico Elevado por hipovolemia Valor predictivo de edema cerebral durante el tratamiento

35
Signos de severidad Estado ácido base Frecuencia respiratoria es proporcional a su severidad KetosisAnion gap Medición de bOHButirato Estado neurológico Compromiso severo al ingreso es factor pronóstico Déficit de fluidos5 – 10% Duración de los síntomas A > tiempo mayor gravedad

36
Tratamiento CAD

37
Tratamiento La KAD severa requiere de manejo en una unidad de cuidados intensivos pediátrica Al comienzo del tratamiento: –Monitorización estricta para prevenir complicaciones Corrección de volemia Administración de insulina Corrección de alteraciones hidroelectrolíticas y ácido base (Na, K, P, Ca, HCO3)
")
38
Déficit de fluidos y electrolitos ESPE/LWPES recomiendan: KAD severa a moderada tiene un déficit de 5 a 10% del volumen. (50 – 100 ml/kg) El déficit debe corregirse en forma lenta y gradual Difícil evaluar clínicamente Elevación de Nureico y Hematocrito orientan a deshidratación severa, su evolución permite evaluar mejoría
 El déficit debe corregirse en forma lenta y gradual Difícil evaluar clínicamente Elevación de Nureico y Hematocrito orientan a deshidratación severa, su evolución permite evaluar mejoría.")
39
Déficit de fluidos y electrolitos Metas iniciales de expansión de volumen: –Restaurar volumen circulante efectivo, al reponer pérdidas de Na y agua (IC – EC) –Restaurar la filtración glomerular para mejorar el clearance de glucosa y ketonas –Minimizar el riesgo de edema cerebral El déficit debe corregirse en forma lenta y gradual
 –Restaurar la filtración glomerular para mejorar el clearance de glucosa y ketonas –Minimizar el riesgo de edema cerebral El déficit debe corregirse en forma lenta y gradual")
40
Déficit de fluidos y electrolitos Un estudio relaciona altos volúmenes de fluidos las primeras horas con edema cerebral. Parece prudente una corrección gradual del déficit, con solución isotónica El volumen inicial con sol isotónica (Na 140 mEq/L) en KAD moderada a severa parece ser 10 ml/kg en la primera hora. Si el volumen circulante efectivo sigue comprometido se podría dar un 2º bolo en la hora siguiente. No dar más de 20 ml/kg en bolos, excepto shock
 en KAD moderada a severa parece ser 10 ml/kg en la primera hora. Si el volumen circulante efectivo sigue comprometido se podría dar un 2º bolo en la hora siguiente. No dar más de 20 ml/kg en bolos, excepto shock.")
41
Déficit de fluidos y electrolitos No dar más de 1,5 a 2 veces el volumen de mantención. (1500 – 1800 ml/m 2 /día) + (70 – 100 ml/kg en dos días) Ej Pac 10 kg sc 0,45 DH severo –Vol mantención 810 ml + 500 ml= 1310 ml/día Pac 30 kg sc 1 DH moderado –Vol mantención 1800 ml + 1050 ml= 2850 ml/día
 + (70 – 100 ml/kg en dos días) Ej Pac 10 kg sc 0,45 DH severo –Vol mantención 810 ml ml= 1310 ml/día Pac 30 kg sc 1 DH moderado –Vol mantención 1800 ml ml= 2850 ml/día.")
42
Déficit de fluidos y electrolitos Solución isotónica se requiere por 4 a 6 horas. Aporte de K 40 mEq/L (KCl 20 – FosfatoK 20) Posteriormente se reduce concentración de sodio a 70 mEq/L. Evaluar aumento de sodio según lo esperado Evaluación continua de estado mental y de hidratación Reevaluar cuando ketoacidosis ha mejorado y se puede ingesta oral, entonces liberar la administración de fluidos (48 – 72 horas). No más de 3500 ml/m2/dia iv + oral
 Posteriormente se reduce concentración de sodio a 70 mEq/L. Evaluar aumento de sodio según lo esperado Evaluación continua de estado mental y de hidratación Reevaluar cuando ketoacidosis ha mejorado y se puede ingesta oral, entonces liberar la administración de fluidos (48 – 72 horas). No más de 3500 ml/m2/dia iv + oral.")
43
Insulina cristalina Alcanza steady state a los 60 min de infusión (100 – 200 micro u/ml) –Supresión producción de glucosa y cetonas –Estimula metabolismo periférico de glucosa y ketonas Reducción de glucosa –50 – 100 mg/dL/hr No debe reducirse la BIC hasta la mejoría de la cetoacidosis No dar bolos Iniciar infusión por lo menos después de la 1º hora de hidratación BIC: 0,1 – 0,05 u/kg/hr (niños más sensibles a insulina) La concentración debe ser lo mayor posible, ideal 1 u/ml Lo más cerca de la vía venosa (se adhiere a las paredes de la jeringa) No mejora ketosis en 2 – 4 horas: infección, deshidratación, acidosis, insulina mal preparada o adherida No mejora ketosis en 2 – 4 horas: infección, deshidratación, acidosis, insulina mal preparada o adherida
 –Supresión producción de glucosa y cetonas –Estimula metabolismo periférico de glucosa y ketonas Reducción de glucosa –50 – 100 mg/dL/hr No debe reducirse la BIC hasta la mejoría de la cetoacidosis No dar bolos Iniciar infusión por lo menos después de la 1º hora de hidratación BIC: 0,1 – 0,05 u/kg/hr (niños más sensibles a insulina) La concentración debe ser lo mayor posible, ideal 1 u/ml Lo más cerca de la vía venosa (se adhiere a las paredes de la jeringa) No mejora ketosis en 2 – 4 horas: infección, deshidratación, acidosis, insulina mal preparada o adherida No mejora ketosis en 2 – 4 horas: infección, deshidratación, acidosis, insulina mal preparada o adherida")
44
Insulina cristalina BIC No reducir si glicemia 250 a 300 mg/dl: –Cambiar solución a SG5% + Na 70 mEq/L + K según niveles de K y P Si glicemia < 250 mg/dl antes de la mejoría de ketoacidosis, –prevención de hipoglicemia –Aumentar concentración de glucosado a 10 ó 12,5% Glicemias aceptables: – 150 a 200 mg/dL en niños menores – 100 a 150 mg/dL en mayores

45
Insulina cristalina BIC Cuando discontinuar? Anión GAP < 12 ± 2 mmol/L = mEq/L BOHB < 1 mmol/L ó 10,4 mg/dL en dos mediciones consecutivas pH > 7,3 ó HCO3 > 15 mEq/L Glicemia < 200 mgr% Buena tolerancia alimentaria Dar primera dosis sbc de insulina con intervalo de anticipación De acción rápida: 15 minutos antes (Lispro) Regular o corta acción: 30 a 60 minutos antes de la suspensión de BIC Justo antes de la alimentación
 Regular o corta acción: 30 a 60 minutos antes de la suspensión de BIC Justo antes de la alimentación.")
46
Tiempo e n los qu e trabajó Mejoría hiperglicemia e hidratación Movimiento de agua desde LEC al LIC Natremia osmolaridad Natremia Natremia

47
Sodio: control horario por 3 – 4 horas Na debe subir 2,4 meq/L cada 100 mg% glicemia Na= Na medido + (ΔSG/42) Falla en el aumento de la natremia puede ser un signo precoz de riesgo de edema cerebral Aumentar sodio o disminuir velocidad de infusión
 Falla en el aumento de la natremia puede ser un signo precoz de riesgo de edema cerebral Aumentar sodio o disminuir velocidad de infusión")
48
Potasio Aporte de 40 meq/L de solución Si paciente hipokalémico, el potasio debe reponerse con inicio de hidratación Si se encuentra normocalémico debería reponerse con el inicio de la insulina Si hipercalemia, se repondrá cuando llegue a niveles normales

49
ANION GAP NORMAL/ACIDOSIS e n los qu e trabajó Insulina detiene la producción de cetonas Aumenta la producción de bicarbonato Mejoría de acidosis Promueve metabolismo de las cetonas Mejora acidosis Mejoría de la perfusión, disminuye lactacidosis Niveles de BOHB < 1 mmol/L

50
Bicarbonato RIESGOS: pCO 2 que atraviesa BHE y genera acidosis cerebral Enlentece ketosis, estimula ketogenesis Factor de riesgo de edema cerebral Hiperosmolaridad Hay evidencia sustancial que APOYA SU NO USO Estudios en adultos y niños no han demostrado su utilidad ADA/ESPE/LWPES Sería beneficioso usado con precaución en –pH< 6,9, con disfunción miocárdica/ vasodilatación –Hipercalemia severa –1 – 2 mmol/kg en 1 a 2 horas.

51
Guías Minsal 2013

52
Resumen Monitorización intensiva ELP – Glicemia – pH – Nu – Hto – CC iniciales HGT horario por 6 horas ELP - pH: horarios por 4 horas, luego cada 2 SF 10 ml/kg en 1 hora máx por 2 veces, luego volumen no mayor de 2 veces el volumen basal (déficit en 48 horas) Control estricto de ascenso de natremia Monitorización de signos de edema cerebral Reposición de K, P No usar bicarbonato
 Control estricto de ascenso de natremia Monitorización de signos de edema cerebral Reposición de K, P No usar bicarbonato")
53
Complicaciones

54
Edema cerebral Disfunción neurocognitiva Trombosis venosa (CVC femoral) Aspiración Arritmias (P, K) Elevación enzimas pancreáticas (40% KAD)
 Aspiración Arritmias (P, K) Elevación enzimas pancreáticas (40% KAD)")
55
Edema cerebral Causas no bien definidas Prevención de edema cerebral: prev KAD 4 – 12 horas terapia 1% de los episodios de KAD Mortalidad 20 – 90% 15 – 20% secuelados Síntomas aparecen con la terapia. 20% ocurre pretto Estudio 41 niños KAD –56% presentan estrechamiento ventricular en RM que disminuye durante recuperación. –60% GCS < 15

56
Edema cerebral: Mecanismos Isquemia/ edema citotóxico: RM : disminución de N acetil aspartato Aumento niveles de lactato en gg basales Edema vasogénico/Alteración de BHE RM: difusión de agua a cerebro Flujo cerebral normal o aumentado a pesar de hipocapnia Edema osmótico por fluidoterapia

57
Edema cerebral: Factores de Riesgo < edad Debut de DM tipo I Elevación de natremia distinta a lo esperado, sugerente de > disminución de osmolaridad –(> 1 – 2 mosm/kg en 2 horas) N ureico elevado al momento del diagnóstico Severidad de la acidosis al diagnóstico Administración de bicarbonato pCO2 inicial más baja
 N ureico elevado al momento del diagnóstico Severidad de la acidosis al diagnóstico Administración de bicarbonato pCO2 inicial más baja")
58
Edema cerebral: Criterios sospecha MENORES Vómitos Cefalea Letargia Pd> 90 mmHg Edad < 5 años MAYORES Nivel de conciencia alterado/fluctuante Desaceleración de FC (> 20 latidos por minuto) Incontinencia no acorde a la edad La presencia de 2 criterios mayores ó 1 mayor más 1 menor indican alta sospecha
 Incontinencia no acorde a la edad La presencia de 2 criterios mayores ó 1 mayor más 1 menor indican alta sospecha")
59
Edema cerebral: Criterios diagnósticos Respuesta motora o verbal al dolor anormales Postura de decorticación o descerebración Parálisis de nervios III, IV y VI Pattern respiratorio anormal

60
Edema cerebral: Tratamiento Intubación y VM Evitar hiperventilación Evitar pCO 2 < 22 mmHg Si se sospecha, iniciar terapia precoz Disminuir velocidad de fluidoterapia Manitol 0,25 – 1 gr/kg en 20 minutos NaCl 3% 5 – 10 ml/kg en 30 minutos

61
Resumen Edema cerebral responsable del 50 a 80% de las muertes por KAD 20% de mortalidad / 15 – 35% secuelas Grupos de riesgo Importante la evaluación clínica. TAC es tardío Sospecha de edema debe tratarse VM en forma cuidadosa

62
Bibliografía European Society for Pediatric Endocrinology/Lawson Wilkins Pediatric Endocrine SocietyESPE/LWPES 2004 American Diabetes Association ADA 2006 International Society for Pediatric and Adolescent Diabetes ISPAD 2014 UpToDateSeptiembre 2014

63
GRACIAS

Presentaciones similares