Descargar la presentación
La descarga está en progreso. Por favor, espere

1
GUILLAIN BARRE Y CIDP DRA. NELLY LUZA
RESIDENTE 1° AÑO NEUROLOGÍA PEDIÁTRICA Universidad de chile, HSBA TUTORA: DRA. Verónica sáez julio2015

2
En 1859 introduce el término Parálisis Aguda Ascendente
A principio del siglo XX, durante la primera Guerra Mundial, Georges Guillainy Jean-Alexandre Barré médicos del ejército francés conocen a dos soldados que padecieron una parálisis parcial y posteriormente se recuperaron de este trastorno. En 1916, Guillain, Barré y Strohl quien llevó a cabo los estudios electrofisiológicos, publican su clásico informe sobre este trastorno e identifican el aumento en la concentración de proteínas en el líquido cefalorraquídeo (LCR) sin elevación en el número de células (disociación albúmino-citológica). En 1916, publican un informe sobre este trastorno e identifican el aumento en la concentración de proteínas en el líquido cefalorraquídeo sin elevación en el número de células (disociación albúmino-citológica).
 sin elevación en el número de células (disociación albúmino-citológica). En 1916, publican un informe sobre este trastorno e identifican el aumento en la concentración de proteínas en el líquido cefalorraquídeo sin elevación en el número de células (disociación albúmino-citológica).")
3
Generalidades Sindrome de Guillain Barre se define como una poliradiculoneuropatía caracterizada por una parálisis arrefléctica. Tiene una incidencia de alrededor de 1-2 por niños menores de 18 años y 1,6 varones afectados por cada mujer. La incidencia es menor en niños que en adultos y por cada 10 años de vida posterior a la primera década de vida, aumenta en 20%. Es raro en menores de 2 años Enfermedad autoinmune frecuentemente gatillada por una infección precedente. En alrededor del 65% de los casos existe un antecedente de infección entre 3 días y dos semanas previo al inicio de la desmielinización.

4
Microorganismos asociados
Campylobacter (demostrado en 30% de los casos) Micoplasma Citomegalovirus Virus de Epstein Barr virus coxsackie y echo. VIH Inmunización Varicela zoster Sarampión Parotiditis Hepatitis A y B Rubéola, influenza A y B Cirugía Trauma Transplante de médula ósea Los microorganismos involucrados son múltiples. asociación en forma con Campylobacter, Micoplasma, citomegalovirus, virus de Epstein Barr, virus vacuna, probable con varicela zoster, sarampión, parotiditis, hepatitis A y B y posible con rubéola, influenza A y B y virus coxsackie y echo. Más recientemente se ha implicado el virus de inmunodeficiencia humana. Se ha demostrado en estudios epidemiológicos que el riesgo de desarrollar un SGB aumenta veces en los 2 meses siguientes a una infección sintomática con Campylobacter.
 Micoplasma. Citomegalovirus. Virus de Epstein Barr. virus coxsackie y echo. VIH. Inmunización. Varicela zoster. Sarampión. Parotiditis. Hepatitis A y B. Rubéola, influenza A y B. Cirugía. Trauma. Transplante de médula ósea. Los microorganismos involucrados son múltiples. asociación en forma con Campylobacter, Micoplasma, citomegalovirus, virus de Epstein Barr, virus vacuna, probable con varicela zoster, sarampión, parotiditis, hepatitis A y B y posible con rubéola, influenza A y B y virus coxsackie y echo. Más recientemente se ha implicado el virus de inmunodeficiencia humana. Se ha demostrado en estudios epidemiológicos que el riesgo de desarrollar un SGB aumenta 100 veces en los 2 meses siguientes a una infección sintomática con Campylobacter.")
5
Mecanismos fisiopatológicos
Una agresión inmune primaria a superficie de célula de Schwann en la Polineuropatía Inflamatoria Desmielinizante Aguda Agresión inmunológica primaria a epítopos en membrana axonal en las formas axonales. Existen diferencias patológicas, electrodiagnósticas y en ocasiones clínicas entre los distintos subtipos, sin embargo mantienen similitudes clínicas y de líquido cefalorraquídeo (LCR) suficientes como para incluirlos en la denominación de Síndrome de Guillain Barre.
 suficientes como para incluirlos en la denominación de Síndrome de Guillain Barre.")
7
Clasificación I. Desmielinizante Inflamatoria Aguda (AIDP) II. Axonal
Neuropatía Axonal Sensitivo Motora (AMSAN) Neuropatía Axonal Motora (AMAN) III. Sindrome Miller Fisher IV. Patrones Restringidos (asociación con SGB controvertida, pueden ser variantes de AIDP) como síndrome sensitivo puro, pandisautonomía, variantes motoras restringidas y otras. De acuerdo a estudios epidemiológicos, la forma Desmielinizante Inflamatoria Aguda (AIDP) constituye la forma más frecuente en países occidentales, (85 a 90% de los casos), aunque en países como China, México y Perú son mas prevalente las formas axonales. La variante de Miller Fisher, constituye alrededor del 3 a 5% de los casos en occidente.
 Neuropatía Axonal Motora (AMAN) III. Sindrome Miller Fisher. IV. Patrones Restringidos (asociación con SGB controvertida, pueden ser variantes de AIDP) como síndrome sensitivo puro, pandisautonomía, variantes motoras restringidas y otras. De acuerdo a estudios epidemiológicos, la forma Desmielinizante Inflamatoria Aguda (AIDP) constituye la forma más frecuente en países occidentales, (85 a 90% de los casos), aunque en países como China, México y Perú son mas prevalente las formas axonales. La variante de Miller Fisher, constituye alrededor del 3 a 5% de los casos en occidente.")
9
Forma Desmielinizante Inflamatoria Aguda
Debilidad progresiva en más de un miembro y arreflexia. El grado de compromiso variable (mínima debilidad en piernas parálisis de extremidades, tronco y pares craneanos, incluyendo parálisis bulbar, facial y oftalmoplejia). En niños es frecuente el inicio con dolor o parestesias en extremidades inferiores (tobillos y dedos), se rehúsan a caminar por el dolor o presentan dificultad. Se produce una progresión ascendente de la debilidad (2 semanas en el 75% de los casos, 3 semanas en el 80% y 4 semanas en más del 90% de los casos) Puede presentarse inestabilidad vasomotora, hipertensión, arritmias cardíacas y secreción inapropiada de hormona antidiurética. Alrededor del 5 % de los pacientes presentan oftalmoparesia, se puede confundir con una miastenia o botulismo Puede haber edema de papila en menos del 5%. The classic presentation of GBS begins with fine paresthesias in the toes and fingertips followed by lower extremity symmetric or modestly asymmetric weakness that may ascend over hours to days to involve the arms and, in severe cases, the muscles of respiration [11,12]. The predominant symptoms of GBS at presentation in children are pain and gait difficulty [13]. In preschool-aged children, the most common symptoms are refusal to walk and pain in the legs [14]. In a prospective series of 95 children with GBS, the most frequent initial symptoms were gait unsteadiness, neuropathic pain, and inability to walk, occurring in 45, 34, and 24 percent, respectively [15]. By the peak of the illness, the frequency of symptoms was as follows: ●79 percent had neuropathic pain ●60 percent could not walk ●51 percent had autonomic dysfunction ●46 percent had cranial nerve involvement ●24 percent could not use their arms ●13 percent required mechanical ventilation
. En niños es frecuente el inicio con dolor o parestesias en extremidades inferiores (tobillos y dedos), se rehúsan a caminar por el dolor o presentan dificultad. Se produce una progresión ascendente de la debilidad (2 semanas en el 75% de los casos, 3 semanas en el 80% y 4 semanas en más del 90% de los casos) Puede presentarse inestabilidad vasomotora, hipertensión, arritmias cardíacas y secreción inapropiada de hormona antidiurética. Alrededor del 5 % de los pacientes presentan oftalmoparesia, se puede confundir con una miastenia o botulismo. Puede haber edema de papila en menos del 5%. The classic presentation of GBS begins with fine paresthesias in the toes and fingertips followed by lower extremity symmetric or modestly asymmetric weakness that may ascend over hours to days to involve the arms and, in severe cases, the muscles of respiration [11,12]. The predominant symptoms of GBS at presentation in children are pain and gait difficulty [13]. In preschool-aged children, the most common symptoms are refusal to walk and pain in the legs [14]. In a prospective series of 95 children with GBS, the most frequent initial symptoms were gait unsteadiness, neuropathic pain, and inability to walk, occurring in 45, 34, and 24 percent, respectively [15]. By the peak of the illness, the frequency of symptoms was as follows: ●79 percent had neuropathic pain. ●60 percent could not walk. ●51 percent had autonomic dysfunction. ●46 percent had cranial nerve involvement. ●24 percent could not use their arms. ●13 percent required mechanical ventilation.")
11
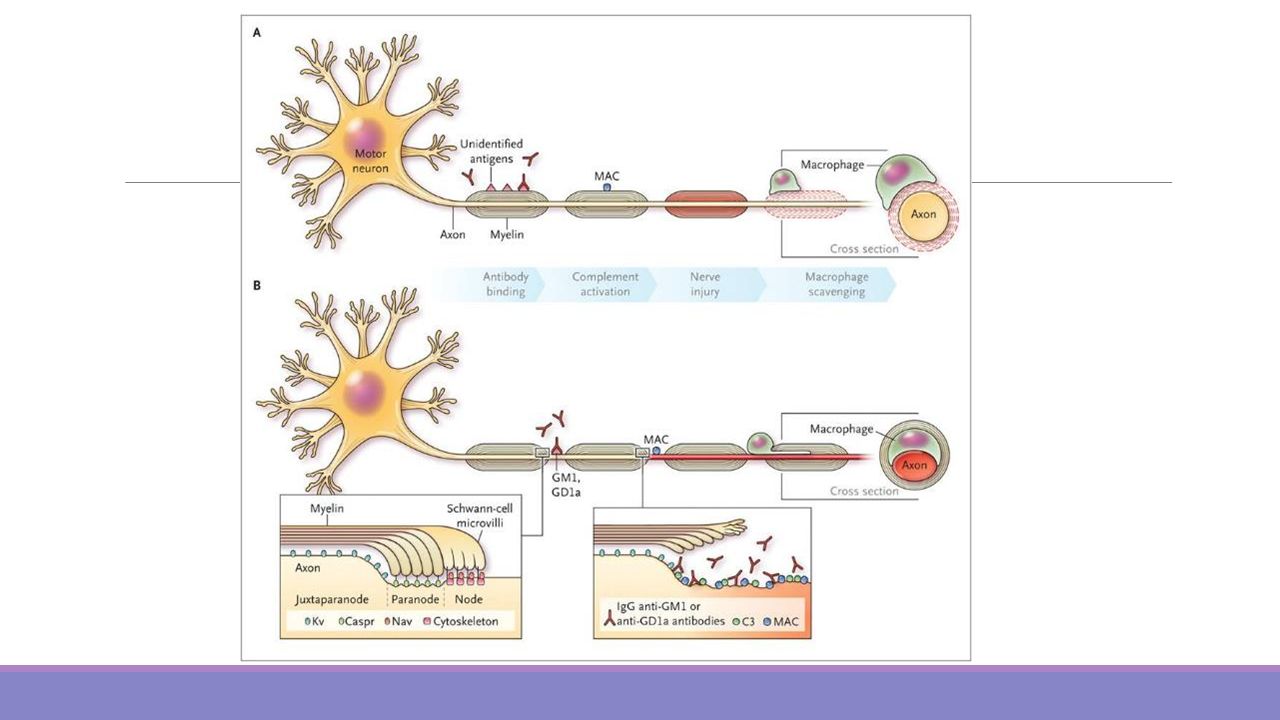
Activación del complemento en la superficie de la célula de Schwann.
Fisiopatología Existe inflamación focal y difusa con infiltración linfocítica y de macrófagos afectando fibras sensitivas y motoras, con mayor compromiso de raíces y plexos proximales adyacentes. Activación del complemento en la superficie de la célula de Schwann. Adherencia de anticuerpos a epítopos en la membrana mas externa de la célula de Schwann. Apertura poros, entrada de calcio a la célula y activación de enzimas proteasas y fosfolipasas que degradan las proteínas de la mielina. Lipopolisacáridos aislados de Campilobacter Jejuni comparten epítopos glicoconjugados con el nervio periférico, incluyendo GM1, GQ1b y GD1a.

12
El curso clínico en niños es más corto que en adultos y la recuperación es más completa (promedio de 43 a 52 días comparado con 85 días)
")
13
Exámenes LCR: elevación de la concentración de proteínas en el líquido cefalorraquídeo sin aumento de celularidad (< 10 células por mm3). Estudio electrofisiológico: Se considera indispensable para el diagnóstico, ya que tiene implicancias en el tratamiento y pronóstico. Los signos electrofisiológicos más precoces son la ausencia de reflejo H, la disminución en la amplitud de los potenciales musculares evocados por electroestimulación (CMAP) y bloqueo de conducción. Posteriormente se produce enlentecimiento de la velocidad de conducción mayor en segmentos proximales con prolongación de la latencia de la onda F. La presencia de fibrilaciones y ondas positivas se correlaciona con compromiso axonal. Resonancia magnética: con gadolinio puede demostrar refuerzo de raíces o nervios periféricos en algunos pacientes, pero estos hallazgos no son específicos y no se requieren para el diagnóstico.
 y bloqueo de conducción. Posteriormente se produce enlentecimiento de la velocidad de conducción mayor en segmentos proximales con prolongación de la latencia de la onda F. La presencia de fibrilaciones y ondas positivas se correlaciona con compromiso axonal. Resonancia magnética: con gadolinio puede demostrar refuerzo de raíces o nervios periféricos en algunos pacientes, pero estos hallazgos no son específicos y no se requieren para el diagnóstico.")
14
Criterios de desmielinización Delanoe y cols.
Presencia de al menos 4 de los siguientes criterios en 3 nervios (deben ser al menos 2 nervios motores y uno sensitivo): 1. Reducción de la velocidad de conducción motora: a. Menor del 80% del límite bajo de lo normal (LBN) si la amplitud es mayor del 80% del LBN. b. Menor del 70% del LBN si la amplitud es menor del 80% del LBN. 2. Bloqueo parcial de la conducción: menos del 15% de cambio en la duración del potencial evocado motor (PEM), entre la estimulación proximal y distal y más del 20% de disminución en la amplitud de pico a pico (o del área negativa) entre el estímulo proximal y distal. 3. Dispersión temporal: Más del 15% de cambio en la duración del potencial entre la estimulación proximal y distal 4. Latencias distales motoras prolongadas: a. Latencia mayor que el 125% del límite alto de la normalidad (LAN) si la amplitud del PEM es mayor del 80% del LBN. b. Latencia mayor del 150% del LAN si la amplitud del PEM es menor del 80% del LBN. 5. Ausencia de ondas F o aumento de las latencias mínimas de las ondas F (latencia mayor que el 120% del LAN si la amplitud del potencial evocado motor es superior al 80% del LBN). 6. Velocidad de conducción sensitiva: la misma definición que la referida para los nervios motores (punto 1). 7. Disminución de la amplitud del potencial evocado motor (PEM) o del potencial sensitivo, debiendo ser menor del 80% del LBN. Nota: el estudio se realiza mediante estimulación nerviosa con electrodos cutáneos, a intensidad supramáxima, y registrando con electrodos también de superficie.
: 1. Reducción de la velocidad de conducción motora: a. Menor del 80% del límite bajo de lo normal (LBN) si la amplitud es mayor del 80% del LBN. b. Menor del 70% del LBN si la amplitud es menor del 80% del LBN. 2. Bloqueo parcial de la conducción: menos del 15% de cambio en la duración del potencial evocado motor (PEM), entre la estimulación proximal y distal y más del 20% de disminución en la amplitud de pico a pico (o del área negativa) entre el estímulo proximal y distal. 3. Dispersión temporal: Más del 15% de cambio en la duración del potencial entre la estimulación proximal y distal. 4. Latencias distales motoras prolongadas: a. Latencia mayor que el 125% del límite alto de la normalidad (LAN) si la amplitud del PEM es mayor del 80% del LBN. b. Latencia mayor del 150% del LAN si la amplitud del PEM es menor del 80% del LBN. 5. Ausencia de ondas F o aumento de las latencias mínimas de las ondas F (latencia mayor que el 120% del LAN si la amplitud del potencial evocado motor es superior al 80% del LBN). 6. Velocidad de conducción sensitiva: la misma definición que la referida para los nervios motores (punto 1). 7. Disminución de la amplitud del potencial evocado motor (PEM) o del potencial sensitivo, debiendo ser menor del 80% del LBN. Nota: el estudio se realiza mediante estimulación nerviosa con electrodos cutáneos, a intensidad supramáxima, y registrando con electrodos también de superficie.")
15
Criterios diagnósticos

16
Escala funcional de gravedad clínica
0. sano, normal. 1. síntomas y signos leves, pero que le permiten hacer las actividades de andar, correr aún con dificultad, actividades de vestido, comida y aseo. 2. puede caminar más de 5 metros sin ayuda ni apoyo, pero no saltar o realizar actividades para su cuidado personal. 3. puede caminar más de 5 metros pero con ayuda o apoyo. 4. está confinado en cama. 5. con ventilación asistida a tiempo total o parcial. 6. muerte

17
Neuropatía Axonal Motora (AMAN)
Forma epidémica descrita en China Se han ido reconociendo con mayor frecuencia casos esporádicos en países occidentales, especialmente América Latina, donde podrían alcanzar a un 40% de los casos asociada con Campylobacter Jejuni. ausencia de sintomatología sensitiva y reflejos osteotendinosos en ocasiones conservados o incluso hiperactivos. Los pacientes tienden a tener un mejor pronóstico y una recuperación similar a la AIDP (degeneración distal de axones). El tratamiento de elección sería la inmunoglobulina IV, ya que se ha descrito falta de respuesta a la plasmaféresis. Acute motor axonal neuropathy — Acute motor axonal neuropathy (AMAN) is a pure motor form of GBS. This disorder is distinguished from AIDP by its involvement of predominantly motor nerves and an electrophysiologic pattern suggesting axonal damage. AMAN occurs mainly in northern China, but is also a common form of GBS in other locations, including Japan, Mexico, and South America [31-37]. It is more common in developing nations, has a seasonal incidence, and is associated with a preceding Campylobacter jejuni infection The presenting clinical features and recovery are similar to those of AIDP [38]. However, more patients have respiratory failure requiring assisted ventilation.
. El tratamiento de elección sería la inmunoglobulina IV, ya que se ha descrito falta de respuesta a la plasmaféresis. Acute motor axonal neuropathy — Acute motor axonal neuropathy (AMAN) is a pure motor form of GBS. This disorder is distinguished from AIDP by its involvement of predominantly motor nerves and an electrophysiologic pattern suggesting axonal damage. AMAN occurs mainly in northern China, but is also a common form of GBS in other locations, including Japan, Mexico, and South America [31-37]. It is more common in developing nations, has a seasonal incidence, and is associated with a preceding Campylobacter jejuni infection. The presenting clinical features and recovery are similar to those of AIDP [38]. However, more patients have respiratory failure requiring assisted ventilation.")
18
Fisiopatología No presenta inflamación.
El hallazgo principal es la degeneración axonal. El ataque inmune primario en estas formas parece estar dirigido a los nodos motores de Ranvier, exista o no degeneración axonal. 1) activación de complemento 2) adhesión de macrófagos a los nodos 3) apertura de espacios periaxonales 4) migración de macrófagos a los espacios periaxonales 5) contracción axonal consecuente y degeneración axonal.
 activación de complemento. 2) adhesión de macrófagos a los nodos. 3) apertura de espacios periaxonales. 4) migración de macrófagos a los espacios periaxonales. 5) contracción axonal consecuente y degeneración axonal.")
19
Neuropatía Axonal Sensitivo Motora (AMSAN)
Primera descripción del tipo axonal de Guillain Barre en por Feasby et al., generó controversia ya que hasta entonces solo se aceptaba la forma desmielinizante. Frecuente en adultos e infrecuente en niños Con destrucción axonal en fibras motoras y sensitivas Curso prolongado y recuperación lenta y parcial Acute motor-sensory axonal neuropathy (AMSAN) resembles the motor axonal variant but has more sensory symptoms. The course tends to be prolonged [39,40]. The pathology is predominantly axonal lesions of both motor and sensory nerve fibers. This form of GBS is recognized infrequently in children. The infrequent diagnosis may be caused in part by the difficulty of performing sensory testing in children and because electrophysiologic studies often are not done.
 resembles the motor axonal variant but has more sensory symptoms. The course tends to be prolonged [39,40]. The pathology is predominantly axonal lesions of both motor and sensory nerve fibers. This form of GBS is recognized infrequently in children. The infrequent diagnosis may be caused in part by the difficulty of performing sensory testing in children and because electrophysiologic studies often are not done.")
20
Síndrome de Miller Fisher
Descrito en por C. Miller Fisher Ataxia de extremidades Arreflexia Oftalmoplejia externa 5% de los casos en adultos y 1% de los casos en edad pediátrica Mas del 90% de los casos tienen anticuerpos IgG antigangliósido anti -GQ1b Miller Fisher syndrome — Miller Fisher syndrome (MFS) is characterized by external ophthalmoplegia, ataxia, and muscle weakness with areflexia [41,42]. Incomplete forms include acute ophthalmoplegia without ataxia, and acute ataxic neuropathy without ophthalmoplegia [5]. Cerebrospinal fluid findings and electrophysiologic features are similar to those in acute inflammatory demyelinating polyneuropathy. Brainstem auditory evoked potentials demonstrate peripheral and central conduction defects [25].
 is characterized by external ophthalmoplegia, ataxia, and muscle weakness with areflexia [41,42]. Incomplete forms include acute ophthalmoplegia without ataxia, and acute ataxic neuropathy without ophthalmoplegia [5]. Cerebrospinal fluid findings and electrophysiologic features are similar to those in acute inflammatory demyelinating polyneuropathy. Brainstem auditory evoked potentials demonstrate peripheral and central conduction defects [25].")
21
Encefalitis Bickerstaff (encephalopatía e hiperreflexia con oftalmoplegia y ataxiaasociada a Ac. anti-GQ1b, respondedora a Ig y plasmaféresis) Polineuritis craneal (alteracióin aguda y severa de nervios craneales y pérdida sensitive severa, debilidad facial bilateral, disfagia, disfonía con conservación de nervio óptico. Se da en pacientes más jóvenes, con mayor requerimiento de VM, pero con recuperación completa. Asociada a infecciones por citomegalovirus. RM con gadolineo muestra captación de nervios craneales) Debilidad fariengea- cervical- braquial: debilidad aguda con disfunción deglutoria. Representa una forma localizada de patología Axonal. Se asocial a IgG autoanticuarpo para GT1a, GQ1b. Bickerstaff encephalitis — Bickerstaff encephalitis is a brainstem encephalitis characterized by encephalopathy and hyperreflexia with features of MFS such as ophthalmoplegia and ataxia. It is not only clinically linked to MFS, but is associated with anti-GQ1b antibodies and can respond to IVIG and plasma exchange [43,44]. Some experts consider MFS Bickerstaff encephalitis, and pharyngeal-cervical-brachial weakness with anti-GQ1b antibodies to be overlapping expressions of the anti-GQ1b antibody syndrome [45]. Polyneuritis cranialis — Patients with polyneuritis cranialis develop acute bilateral multiple cranial nerve involvement and severe peripheral sensory loss. They typically have bilateral facial weakness, dysphagia, and dysphonia with optic nerve sparing. Patients tend to be younger than those with other types. This variant is associated with preceding cytomegalovirus infections [46]. Cerebrospinal fluid findings and electrophysiologic features of polyneuritis cranialis are similar to those of AIDP. MRI with gadolinium shows enhancement of multiple cranial nerves [47]. More children with this variant require ventilator support than those with the more typical presentation of GBS [48]. However, most recover fully. Pharyngeal-cervical-brachial weakness — The pharyngeal-cervical-brachial variant of GBS is characterized by acute weakness of the oropharyngeal, neck, and shoulder muscles with swallowing dysfunction [49-51]. Facial weakness may be present as well. Leg strength and leg reflexes are usually preserved. This form may overlap with Miller Fisher syndrome [50,52]. It is thought to represent a localized form of axonal GBS [5,49,50]. Some patients with pharyngeal-cervical-brachial weakness have IgG autoantibodies to GT1a, GQ1b, or less often to GD1a.
 Debilidad fariengea- cervical- braquial: debilidad aguda con disfunción deglutoria. Representa una forma localizada de patología Axonal. Se asocial a IgG autoanticuarpo para GT1a, GQ1b. Bickerstaff encephalitis — Bickerstaff encephalitis is a brainstem encephalitis characterized by encephalopathy and hyperreflexia with features of MFS such as ophthalmoplegia and ataxia. It is not only clinically linked to MFS, but is associated with anti-GQ1b antibodies and can respond to IVIG and plasma exchange [43,44]. Some experts consider MFS Bickerstaff encephalitis, and pharyngeal-cervical-brachial weakness with anti-GQ1b antibodies to be overlapping expressions of the anti-GQ1b antibody syndrome [45]. Polyneuritis cranialis — Patients with polyneuritis cranialis develop acute bilateral multiple cranial nerve involvement and severe peripheral sensory loss. They typically have bilateral facial weakness, dysphagia, and dysphonia with optic nerve sparing. Patients tend to be younger than those with other types. This variant is associated with preceding cytomegalovirus infections [46]. Cerebrospinal fluid findings and electrophysiologic features of polyneuritis cranialis are similar to those of AIDP. MRI with gadolinium shows enhancement of multiple cranial nerves [47]. More children with this variant require ventilator support than those with the more typical presentation of GBS [48]. However, most recover fully. Pharyngeal-cervical-brachial weakness — The pharyngeal-cervical-brachial variant of GBS is characterized by acute weakness of the oropharyngeal, neck, and shoulder muscles with swallowing dysfunction [49-51]. Facial weakness may be present as well. Leg strength and leg reflexes are usually preserved. This form may overlap with Miller Fisher syndrome [50,52]. It is thought to represent a localized form of axonal GBS [5,49,50]. Some patients with pharyngeal-cervical-brachial weakness have IgG autoantibodies to GT1a, GQ1b, or less often to GD1a.")
22
Otras variantes Pandisautonomía Aguda
Forma Sensitiva Pura (ataxia sensitiva, ROT-, alteración motora mínima. Asociación a Ac. GD1b) Diplejia Facial y Parestesia Distal (Variante de AIDP) Parálisis VI Par y Paresthesia Distal Radiculopatía Bilateral Lumbar Paraparesias (debilidad en extremidades inferiores) ●Acute pandisautonomia, which may respond to intravenous immune globulin [53]. Symptoms include diarrhea, vomiting, dizziness, abdominal pain, ileus, orthostatic hypotension, urinary retention, pupillary abnormalities, an invariant heart rate, decreased sweating, salivation, and lacrimation. Reflexes are absent or diminished and sensory symptoms may be present [54]. ●Pure sensory GBS, with involvement of large sensory fibers leading to significant sensory ataxia [55]. Reflexes are absent and there may be minor motor involvement. An association with antibodies to GD1b has been noted. ●Facial diplegia and distal limb paresthesia [52]. This is considered a variant of acute inflammatory demyelinating polyneuropathy [5]. ●Sixth nerve palsy and distal paresthesia [52]. ●Bilateral lumbar radiculopathy [52]. ●Paraparesis, with weakness restricted to the legs at presentation [49,56]. A minority experience some arm weakness over the course of the illness.
 Diplejia Facial y Parestesia Distal (Variante de AIDP) Parálisis VI Par y Paresthesia Distal. Radiculopatía Bilateral Lumbar. Paraparesias (debilidad en extremidades inferiores) ●Acute pandisautonomia, which may respond to intravenous immune globulin [53]. Symptoms include diarrhea, vomiting, dizziness, abdominal pain, ileus, orthostatic hypotension, urinary retention, pupillary abnormalities, an invariant heart rate, decreased sweating, salivation, and lacrimation. Reflexes are absent or diminished and sensory symptoms may be present [54]. ●Pure sensory GBS, with involvement of large sensory fibers leading to significant sensory ataxia [55]. Reflexes are absent and there may be minor motor involvement. An association with antibodies to GD1b has been noted. ●Facial diplegia and distal limb paresthesia [52]. This is considered a variant of acute inflammatory demyelinating polyneuropathy [5]. ●Sixth nerve palsy and distal paresthesia [52]. ●Bilateral lumbar radiculopathy [52]. ●Paraparesis, with weakness restricted to the legs at presentation [49,56]. A minority experience some arm weakness over the course of the illness.")
23
Diagnóstico diferencial
Poliomielitis (virus y/o vacuna) Infecciones virales Polirradiculitis por CMV Encefalomielitis aguda diseminada (compromiso SNC) Poliradiculoneuritis desmielinizante inflamatoria crónica (CIDP) Mielopatía Botulismo Difteria Polirradiculopatía de Lyme Porfiria Miastenia Gravis Dermatomiositis aguda Intoxicación por órgano fosforados poliomielitis (virus polio vacuna) o infecciones virales que determinan cuadros tip o polio. Si hay compromiso del sistema nervioso central considerar la encefalomielitis aguda diseminada y si el curso se prolonga más allá de las 4 semanas la Poliradiculoneuritis desmielinizante inflamatoria crónica (CIDP). En cuadros con compromiso sensitivo o esfinteriano grave o persistente debe descartarse una mielopatía, en primer término una compresión medular. La pérdida temprana de reactividad pupilar debe hacer sospechar un Botulismo. Otros cuadros a considerar incluyen la Difteria, Polirradiculopatía de Lyme, Porfiria (dolor abdominal, convulsiones, psicosis), Miastenia Gravis, Dermatomiositis aguda e intoxicación por órgano fosforados
 Infecciones virales. Polirradiculitis por CMV. Encefalomielitis aguda diseminada (compromiso SNC) Poliradiculoneuritis desmielinizante inflamatoria crónica (CIDP) Mielopatía. Botulismo. Difteria. Polirradiculopatía de Lyme. Porfiria. Miastenia Gravis. Dermatomiositis aguda. Intoxicación por órgano fosforados. poliomielitis (virus polio vacuna) o. infecciones virales que determinan cuadros tip o polio. Si hay compromiso del sistema nervioso central. considerar la encefalomielitis aguda diseminada y si el curso se prolonga más allá de las 4 semanas la. Poliradiculoneuritis desmielinizante inflamatoria crónica (CIDP). En cuadros con compromiso sensitivo. o esfinteriano grave o persistente debe descartarse una mielopatía, en primer término una. compresión medular. La pérdida temprana de reactividad pupilar debe hacer sospechar un Botulismo. Otros cuadros a considerar incluyen la Difteria, Polirradiculopatía de Lyme, Porfiria (dolor abdominal, convulsiones, psicosis), Miastenia Gravis, Dermatomiositis aguda e intoxicación por órgano fosforados.")
24
Manejo, tratamiento y pronóstico
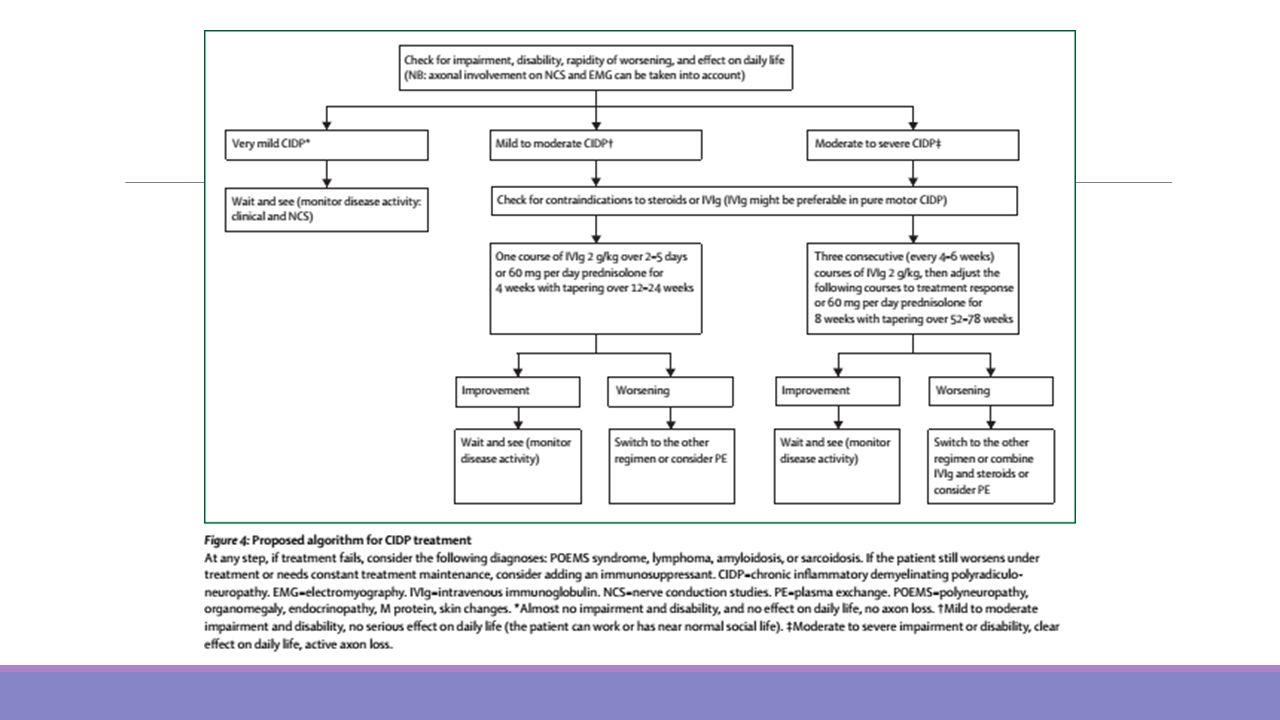
Hospitalización en un centro con UCI, cuidar posicionamiento, prevenir la aparición de contracturas y escaras y asegurar un aporte calórico proteico adecuado. La plasmaféresis y las inmunoglobulinas intravenosas (IgG IV) han demostrado similar eficacia, acelerando la recuperación, aunque no disminuyen la mortalidad. En niños es tratamiento de primera línea la Inmunoglobulina IV en dosis total de 2 gramos/kg de peso, administradas en 2 a 5 días. En el uso de plasmaféresis se recomienda recambio plasmático de 4 sesiones (50ml/kg c/u) para las formas moderadas a graves y 2 sesiones para las formas leves. Se considera adecuado tratar incluso a los pacientes ambulantes pero que no logran caminar más de 5 metros sin asistencia. Los corticoides no mejoran la evolución respecto al placebo. Un 30% de los pacientes requiere ventilación asistida. Un 85% se recupera ad integrum y la mortalidad es de alrededor de un 5 a 15 % . Entre un 5-10% presentarán una Polineuropatía Desmielinizante Inflamatoria Crónica. Todo paciente debe ser hospitalizado en un centro con UCI y experiencia en el manejo de esta patología. La plasmaféresis y las inmunoglobulinas intravenosas (IgG IV) han demostrado similar eficacia, acelerando la recuperación, aunque no disminuyen la mortalidad. En niños, por las dificultades que plantea llevar a cabo una plasmaféresis completa, se acepta como tratamiento de primera línea la Inmunoglobulina IV en dosis total de 2 gramos/kg de peso, administradas en 2 a 5 días. En el uso de plasmaféresis se recomienda recambio plasmático de 4 sesiones (50ml/kg c/u) para las formas moderadas a graves y 2 sesiones para las formas leves. 16 La decisión de tratar con plasmaféresis o inmunoglobulinas depende del estado funcional. Se considera adecuado tratar incluso a los pacientes ambulantes pero que no logran caminar más de 5 metros sin asistencia. Los corticoides no mejoran la evolución respecto al placebo. Otras medidas de importancia incluyen cuidar posicionamiento, prevenir la aparición de contracturas y escaras y asegurar un aporte calórico proteico adecuado. Un 30% de los pacientes requiere ventilación asistida, un 85% se recupera ad integrum y la mortalidad es de alrededor de un 5 a 15 % . Entre un 5-10% presentarán una Polineuropatía Desmielinizante Inflamatoria Crónica
 han demostrado similar eficacia, acelerando la recuperación, aunque no disminuyen la mortalidad. En niños es tratamiento de primera línea la Inmunoglobulina IV en dosis total de 2 gramos/kg de peso, administradas en 2 a 5 días. En el uso de plasmaféresis se recomienda recambio plasmático de 4 sesiones (50ml/kg c/u) para las formas moderadas a graves y 2 sesiones para las formas leves. Se considera adecuado tratar incluso a los pacientes ambulantes pero que no logran caminar más de 5 metros sin asistencia. Los corticoides no mejoran la evolución respecto al placebo. Un 30% de los pacientes requiere ventilación asistida. Un 85% se recupera ad integrum y la mortalidad es de alrededor de un 5 a 15 % . Entre un 5-10% presentarán una Polineuropatía Desmielinizante Inflamatoria Crónica. Todo paciente debe ser hospitalizado en un centro con UCI y experiencia en el manejo de esta. patología. La plasmaféresis y las inmunoglobulinas intravenosas (IgG IV) han demostrado similar. eficacia, acelerando la recuperación, aunque no disminuyen la mortalidad. En niños, por las. dificultades que plantea llevar a cabo una plasmaféresis completa, se acepta como tratamiento de. primera línea la Inmunoglobulina IV en dosis total de 2 gramos/kg de peso, administradas en 2 a 5. días. En el uso de plasmaféresis se recomienda recambio plasmático de 4 sesiones (50ml/kg c/u) para. las formas moderadas a graves y 2 sesiones para las formas leves. 16. La decisión de tratar con plasmaféresis o inmunoglobulinas depende del estado funcional. Se. considera adecuado tratar incluso a los pacientes ambulantes pero que no logran caminar más de 5. metros sin asistencia. Los corticoides no mejoran la evolución respecto al placebo. Otras medidas de importancia incluyen cuidar posicionamiento, prevenir la aparición de contracturas. y escaras y asegurar un aporte calórico proteico adecuado. Un 30% de los pacientes requiere ventilación asistida, un 85% se recupera ad integrum y la mortalidad. es de alrededor de un 5 a 15 % . Entre un 5-10% presentarán una Polineuropatía Desmielinizante. Inflamatoria Crónica.")
25
POLIRRADICULONEUROPATÍA CRÓNICA DESMIELINIZANTE INFLAMATORIA (CIDP )
")
26
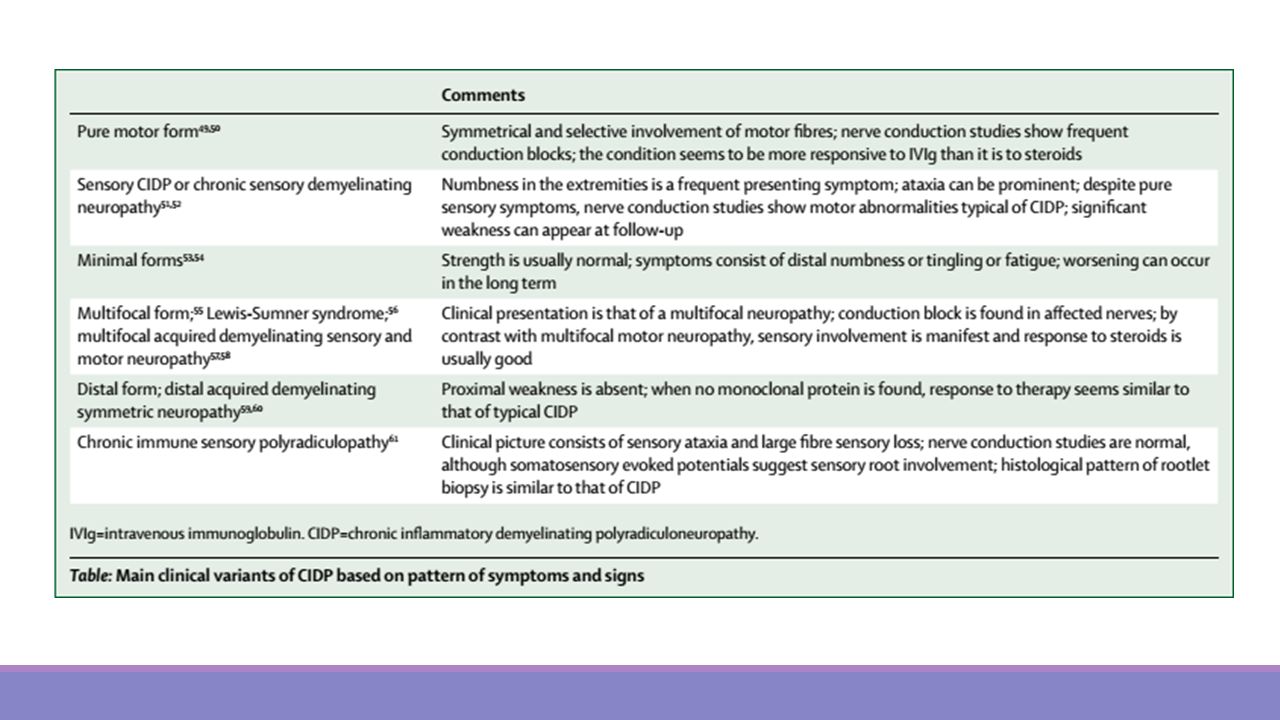
Generalidades Es un cuadro común, probablemente subdiagnosticado y tratable, con una prevalencia estimada de 0,5 por niños y 1 a 2 por adultos. La semejanza clínica con la variedad aguda y los efectos beneficiosos del tratamiento inmunomodulador apuntan a una patogenia inmunomediada. Tiene un espectro de presentación amplio

28
CIDP La forma clásica se manifiesta por debilidad proximal y distal, en ocasiones mayor compromiso proximal, axial y de flexores de cuello con hipo o arreflexia Alteración sensitiva: leve, distal y de predominio en extremidades inferiores, dolorosa o restringida a sensibilidad vibratoria. Puede acompañarse de alteraciones autonómicas (alteraciones miccionales o sindrome de Horner) En algunos casos existe compromiso de sistema nervioso central (papiledema, mielopatía, o ataxia o imagenológico con alteraciones desmielinizantes en neuroeje)
 En algunos casos existe compromiso de sistema nervioso central (papiledema, mielopatía, o ataxia o imagenológico con alteraciones desmielinizantes en neuroeje)")
31
Este cuadro se instala en un período mayor a 8 semanas.
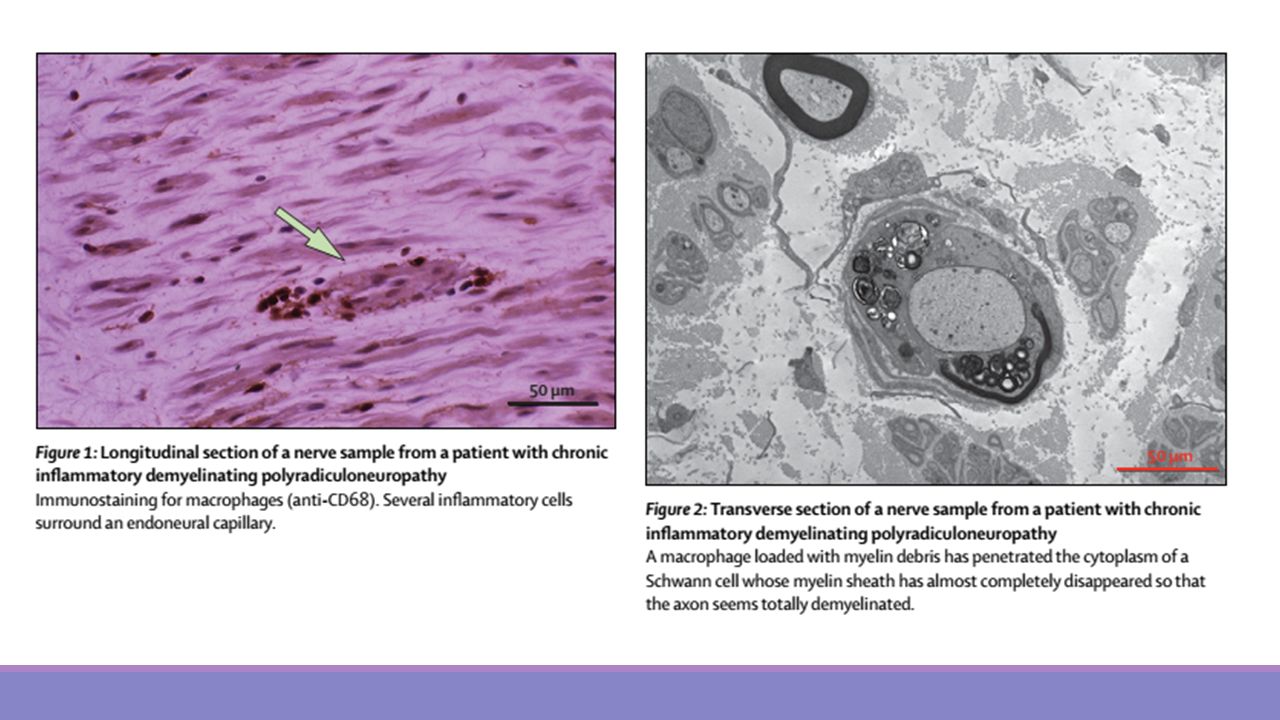
La evolución clínica puede ser crónica recurrente (más frecuente en niños), monofásica o progresiva, en meses o incluso años. Los estudios de conducción muestran desmielinización Existe elevación de proteínas en el LCR. La Resonancia Magnética de médula espinal pueden mostrar captación multirradicular del medio de contraste. El diagnóstico se basa esencialmente en clínica y electrofisiología, siendo el estudio de LCR y la Biopsia de Nervio controvertidos en cuando a su utilidad para el diagnóstico.
, monofásica o progresiva, en meses o incluso años. Los estudios de conducción muestran desmielinización. Existe elevación de proteínas en el LCR. La Resonancia Magnética de médula espinal pueden mostrar captación multirradicular del medio de contraste. El diagnóstico se basa esencialmente en clínica y electrofisiología, siendo el estudio de LCR y la Biopsia de Nervio controvertidos en cuando a su utilidad para el diagnóstico.")
34
Gracias.

35
Bibliografía Cruse, Robert et al.Epidemiology, clinical features, and diagnosis of Guillain-Barré syndrome in children. Up to Date, Nov Avaria y cols. ENFERMEDADES NEUROMUSCULARES. Hospital Roberto del Río Jain-Jim Lin et al. Clinical Variants of Guillain-Barré Syndrome in Children. Pediatric Neurology 47 (2012) 91-96 Jean-Michel Vallat. Chronic inflammatory demyelinating polyradiculoneuropathy: diagnostic and therapeutic challenges for a treatable condition. Lancet Neurology 2010; 9: 402–12 P. David. Neurología Pediátrica. Editoreal Mediterraneo, 2012: S. Pascual. Síndrome de Guillain-Barré. Servicio de Neurología Pediátrica. Hospital Infantil Universitario La Paz, Madrid. Protocolos Pediatría, 2008.
 Jean-Michel Vallat. Chronic inflammatory demyelinating polyradiculoneuropathy: diagnostic and therapeutic challenges for a treatable condition. Lancet Neurology 2010; 9: 402–12. P. David. Neurología Pediátrica. Editoreal Mediterraneo, 2012: S. Pascual. Síndrome de Guillain-Barré. Servicio de Neurología Pediátrica. Hospital Infantil Universitario La Paz, Madrid. Protocolos Pediatría,")
Presentaciones similares